
One step hydrothermal synthesis of Mn3O4/graphene composites with great electrochemical properties for lithium-ion batteries
Abstract
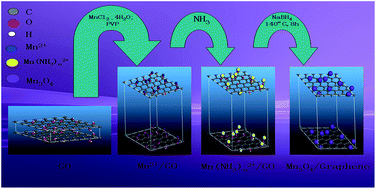
The fabrication and electrochemical performance of Mn3O4/graphene composites are discussed in this work. The main reaction procedures consist of two parts: one is the formation of Mn3O4 particles and the other is the reduction of graphite oxide to graphene. The chemicals, MnCl2·4H2O and NaBH4 are employed as a manganese source and a reduction reagent, respectively. During the formation of Mn3O4 particles, NH3 is added to the reaction system, directly, which simplifies the hydrolysis of amide, and the surfactant, polyvinylpyrrolidone (PVP), is used to ensure great dispersion and size-controlled formation of Mn3O4 particles. The resulting materials are characterized by XRD, SEM, HRTEM, FT-IR, Raman and XPS. Mn3O4 particles dispersing on the surface of graphene have an average diameter of ca. 30 nm. The materials deliver a stable reversible capacity of ca. 500 mA h g−1 at a current density of 60 mA g−1 even after 100 cycles. The reversible capacity of the samples coated with graphene is much better than that of pure materials.
 Please wait while we load your content...
Please wait while we load your content...

