
Pentafluorophenyl vinyl sulfonate enables efficient, metal-free, radical-based alkene hydroacylation with an aldehyde as a limiting reagent†
Abstract
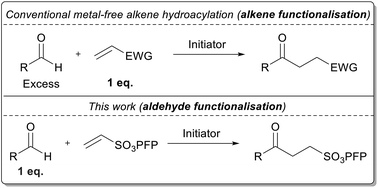
The unique properties of pentafluorophenyl vinyl sulfonate allow for a hitherto unmet feat to be realised; efficient and high yielding, metal-free, radical-based alkene hydroacylation employing aldehyde as limiting reagent. The optimised conditions are shown to work in good yields across a series of aldehydes, thus demonstrating the wide applicability of the developed protocol.
 Please wait while we load your content...
Please wait while we load your content...

