
Simple descriptors for assessing the outcome of aza-Diels–Alder reactions†
Abstract
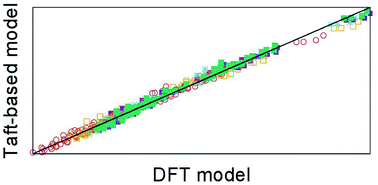
The iminium aza-Diels–Alder (iADA) reaction of cyclopentadiene with 16 protonated alkyl alkylimineglyoxilates was studied using density functional theory (DFT) in order to elucidate how different substitution patterns in the dienophile may affect the reaction’s outcome. Additionally, the application of the polarisable continuum model (PCM) together with the evaluation of the thermodynamic properties at different temperatures further allowed the surveying of the importance of such factors. These effects were combined into linear models which use the temperature, Taft’s constants and characteristics of the solvent as descriptors for modelling and predicting the activation enthalpy and enthalpic balance of the iADA reactions under study. This model performs in a satisfactory manner, providing a cross-validation of the DFT framework traditionally used for predicting the outcome of these reactions and also uncovering novel insights into how the substitution patterns in the dienophile, the solvent and the temperature interact in order to give the characteristic regio- and stereoselectivity of the iADA reaction. Moreover, the results show that Taft’s polar and steric substituent constants are important descriptors for assessing the outcome of iADA reactions.
 Please wait while we load your content...
Please wait while we load your content...

