
Electronic structure and thermoelectric properties of the Zintl compounds Sr5Al2Sb6 and Ca5Al2Sb6: first-principles study†
Abstract
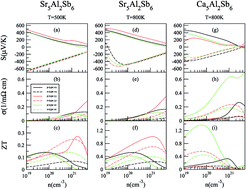
Previous experimental work showed that Zn-doping only slightly increased the carrier concentration of Sr5Al2Sb6 and the electrical conductivity improved barely, which is very different from the results of Zn-doping in Ca5Al2Sb6. To understand their different thermoelectric behaviors, we investigated their stability, electronic structure, and thermoelectric properties using first-principles calculations and the semiclassical Boltzmann theory. We found that the low carrier concentration of Zn-doped Sr5Al2Sb6 mainly comes from its high positive formation energy. Moreover, we predict that a high hole concentration can possibly be realized in Sr5Al2Sb6 by Na or Mn doping, due to the negative and low formation energies of Na- and Mn-doped Sr5Al2Sb6, especially for Mn doping (−6.58 eV). For p-type Sr5Al2Sb6, the large effective mass along Γ–Y induces a large Seebeck coefficient along the y direction, which leads to the good thermoelectric properties along the y direction. For p-type Ca5Al2Sb6, the effective mass along Γ–Z is always smaller than those along the other two directions with increasing doping degree, which induces its good thermoelectric properties along the z direction. The analysis of the weight mobility of the two compounds confirms this idea. The calculated band structure shows that Sr5Al2Sb6 has a larger band gap than Ca5Al2Sb6. The relatively small band gap of Ca5Al2Sb6 mainly results from the appearance of a high density-of-states peak around the conduction band bottom, which originates from the Sb–Sb antibonding states in it.
 Please wait while we load your content...
Please wait while we load your content...

