
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic transfer hydrogenation in γ-valerolactone-based ionic liquids†
Andrea
Strádi
ab,
Márk
Molnár
b,
Péter
Szakál
a,
Gábor
Dibó
bc,
Dániel
Gáspár
a and
László T.
Mika
*a
aBudapest University of Technology and Economics, Department of Chemical and Environmental Process Engineering, Műegyetem rkp. 3, Budapest H-1111, Hungary. E-mail: laszlo.t.mika@mail.bme.hu; Fax: +36 1 463 3197; Tel: +36 1 463 1263
bEötvös Loránd University, Institute of Chemistry, Pázmány Péter str. 1/A, Budapest H-1117, Hungary
cJános Selye University, Faculty of Education, Department of Chemistry, 3322 Bratislavská str., Komárno SK-94501, Slovakia
First published on 12th August 2015
Abstract
The combination of transfer hydrogenation reaction with the advantages of γ-valerolactone-based ionic liquids could result in an environmentally benign method for the reduction of organic substrates. Ionic liquids containing 4-hydroxyvalerate anion were applied as alternative solvents for the reduction of acetophenone, its substituted forms and different alkenes using transition metal based catalysts. The optimal conditions (e.g. type of catalyst precursor and hydrogen donor) for the transformation were also specified.
Introduction
Typically, the commonly used organic solvents are toxic, have high vapour pressure and negative impact on the environment. Their replacement represents a crucial part in the development of greener and environmentally benign chemical technologies,1 and preference should be given to solvents with no or low toxicity according to the fifth principle of green chemistry.1b Recently several alternative solvents e.g. water,2 fluorous solvents,3 supercritical carbon-dioxide,4 alcohols,5 and ionic liquids (ILs) were introduced with significantly different solvent properties.6 Due to their attractive properties, ILs have attracted considerable attention as environmentally benign reaction media. By the proper variation of their anionic and cationic parts, their physical properties such as vapour pressure, thermal stability, solvation strength can easily be fine-tuned.7 Accordingly, wide range of chemical transformations has been successfully performed in room temperature ionic liquids (RTILs),8 including industrially important reactions e.g. Friedel–Crafts acylation,9 hydroformylation,10 and Beckmann-rearrangement.11Hydrogenation reaction as one of the most important and widely studied transition metal catalyzed homogeneous transformation12 was also performed in ionic liquids.13,14 The catalytic transfer hydrogenation of organic substrates is a safer and more energy-efficient alternative operation without using hydrogen gas and can generally be performed in common organic solvents.15 Although, the ionic liquids are widely used as environmentally benign solvents in high-pressure catalysis, there are only a few examples for application in transfer hydrogenation reactions. The combination of the mild conditions of transfer hydrogenation with the advantages of γ-valerolactone (GVL)-based ILs could result in an environment-friendly method for the reduction of organic substrates. In addition, due to the low vapour pressure of GVL based ILs,14 the risk of environmental emission, therefore the potential hazard to health, could be significantly reduced compared to conventional organic solvents.
First, Berthold et al. published the application of ILs as reaction media for transfer hydrogenation of ketones and their α,β-unsaturated forms.16 Hermecz et al. reported the reduction of α,β-unsaturated carbonyl derivatives by using [ClRh(PPh3)3] and [Rh(cod)Cl]2 precursors in imidazolium, ammonium, and phosphonium containing ILs, representing outstanding chemoselectivities depending on the nature of the anions, with TOF = 2.5–20 h−1 at 90 °C.17 Since, the asymmetric hydrogenation of ketones is a key reaction in the synthesis of biologically active molecules, enantioselective transfer hydrogenation reactions were also performed in ILs resulting in enantiomeric excess of 76–96%.18 Dyson19 and Ohta18 revealed that ILs do not act only as solvents, they have a special role in the stabilization of the catalytically active species. Chakraborti et al. proved that ionic liquids could have a special role in the activation of organic substrates e.g. ketones and aldehydes via hydrogen bond formation between the IL and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group.20,21
O group.20,21
It was recently demonstrated, that selective catalytic hydrogenation can be performed in GVL-based ILs.14 This generation of ILs can easily be prepared from bio-originated GVL,14,22 which is a proposed sustainable platform molecule.23 The reaction rate could be affected by varying the substituents of the valerate anion and the tetraalkylammonium cation.14 We also reported that various functionalized olefins can be selectively reduced in tetrabutylammonium 4-hydroxyvalerate [TBA][HV] ionic liquid with TOF = 20–41 h−1. Accordingly, [TBA][HV] was selected as solvent for the transfer hydrogenation of acetophenone, a typical model substrate.
We report here the application of GVL-based ILs as reaction media for transfer hydrogenation reactions using various transition metal catalysts.
Initially, to extend the series of GVL-based ionic liquids, novel species were synthesized as follows: tetraethylammonium 4-hydroxyvalerate ([TEA][HV]), tetrapropylammonium 4-hydroxyvalerate ([TPrA][HV]), and tetrapentylammonium 4-hydroxyvalerate ([TEA][HV]). Since the temperature dependence of the vapour pressure of solvents is a key property, it was determined for novel ILs in the temperature range of 0–85 °C. Expectedly, the data obtained was found to be in the same range as measured in case of GVL and the previously published GVL-based ILs14 as well (Fig. 1).
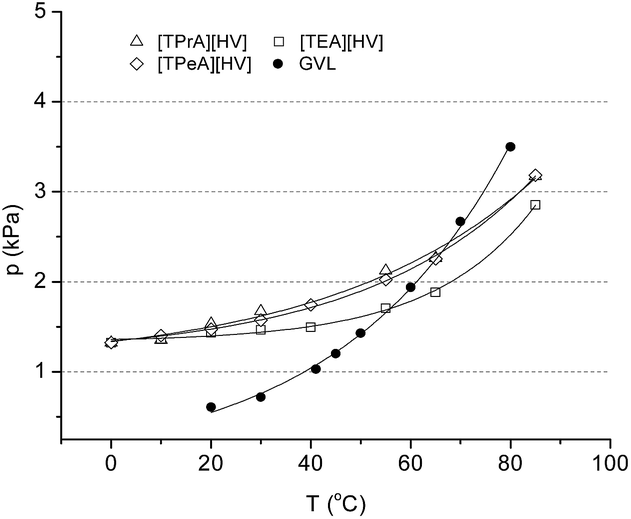 | ||
| Fig. 1 Vapour pressure of GVL-based ILs compared to data for GVL (ref. 21). | ||
Furthermore, different catalyst precursors were investigated in the presence of [TBA][HV] and formic acid as hydrogen donor in the transfer hydrogenation of acetophenone at 80 °C (Scheme 1). Expectedly, in the absence of any catalyst no conversion was detected. The activities of the Rh-based catalyst precursors were much higher than that of obtained for the Pd-containing system (Table 1), in accordance with previous results.17a,24 The activity of the Shvo catalyst, which was found to be very efficient for the conversion of levulinic acid to GVL,25 was rather low under these conditions (Table 1, entry 7). The highest activity was achieved in the case of using [Rh(cod)2]+[BF4]− similarly to the conventional hydrogenation reactions.14 The resulted TOF was 20 h−1 which is significant, compared to literature data reported so far.17 Consequently, further experiments were performed using [Rh(cod)2]+[BF4]−.
 | ||
| Scheme 1 | ||
| Entry | Catalyst precursor | Conv. (%) | TON (–) | TOF (h−1) |
|---|---|---|---|---|
| a Reaction conditions: 0.005 mmol catalyst, 0.3 mmol acetophenone, 1.6 mmol HCOOH, in 0.5 mL [TBA][HV], T = 80 °C, t = 3 h. b {[2,3,4,5-(C6H5)4(η5-C4CO)]2H}Ru2(CO)4(μ-H). | ||||
| 1 | ClRh(PPh3)3 | 92 | 55 | 18.3 |
| 2 | [Rh(cod)Cl]2 | 94 | 56 | 18.6 |
| 3 | [Rh(cod)2]+[BF4]− | >99 | 60 | 20.0 |
| 4 | [Rh(cod)2]+[SbF6]− | 97 | 58 | 19.3 |
| 5 | [Rh(cod)2]+[B(Ph(CF3)2)4]− | 87 | 52 | 17.3 |
| 6 | Pd(OAc)2 | 39 | 23 | 7.6 |
| 7 | Shvo catalystb | 35 | 21 | 7.0 |
In contrast with the conventional high-pressure hydrogenation system, the reaction rate could not be increased by the addition of phosphine ligand. When 10 eq. triphenyl phosphine was added to the reaction mixture, the conversion decreased to 58% under identical conditions. In the presence of the sodium salt of trisulfonated triphenylphosphine (TPPTS), even lower yield (6%) of 1-phenylethanol was obtained. Similar effect was reported for the Pd-catalyzed selective reduction of alkenes.26
To optimize the reaction temperature, we repeated the reduction of acetophenone in [TBA][HV] in the temperature range of 60–90 °C. The highest conversion was obtained at 80 °C (Fig. 2).
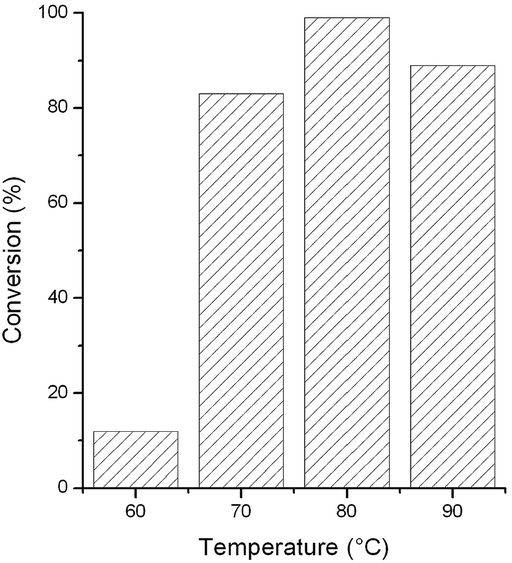 | ||
| Fig. 2 Effect of reaction temperature on the reduction of acetophenone. Reaction conditions: 0.005 mmol [Rh(cod)2]+[BF4]−, 0.3 mmol acetophenone, 1.6 mmol HCOOH, 0.5 mL [TBA][HV], t = 3 h. | ||
The catalytic performance of [Rh(cod)2]+[BF4]− was also evaluated utilizing different conventional hydrogen donors (Table 2). In the presence of formic acid and sodium formate >99% conversion was obtained, while by using potassium formate, the hydrogenation was incomplete. It should be noted that no ester formation was detected in the presence of HCOOH. No product formation could be detected after 3 hours by using ammonium formate (which was not even soluble under the applied conditions) and isopropyl alcohol. In accordance with the 7th principle of green chemistry, formic acid was selected as hydrogen donor since it can be produced from renewable raw materials;27 moreover the carbon dioxide produced in the reaction is thermodynamically very stable and can be easily removed from the reaction mixture.
| Entry | Hydrogen donor | Conversion (%) | TOF (h−1) |
|---|---|---|---|
| a Reaction conditions: 0.005 mmol [Rh(cod)2]+[BF4]−, 0.3 mmol acetophenone, 1.6 mmol H-donor, 0.5 mL [TBA][HV], T = 80 °C, t = 3 h. | |||
| 1 | HCOOH | >99 | 20.0 |
| 2 | HCOONa | >99 | 20.0 |
| 3 | HCOOK | 89 | 17.8 |
| 4 | HCOONH4 | 0 | 0 |
| 5 | iPrOH | 0 | 0 |
It was shown that the reaction rate of the transfer hydrogenation is determined by the concentration of the H-donor.28 The negative effect of acidity on reaction rate was clearly demonstrated in case of asymmetric transfer hydrogenations.29 Accordingly, we investigated this effect in the range of 1.2–6.5 molar excess of HCOOH relative to the substrate (Fig. 3).
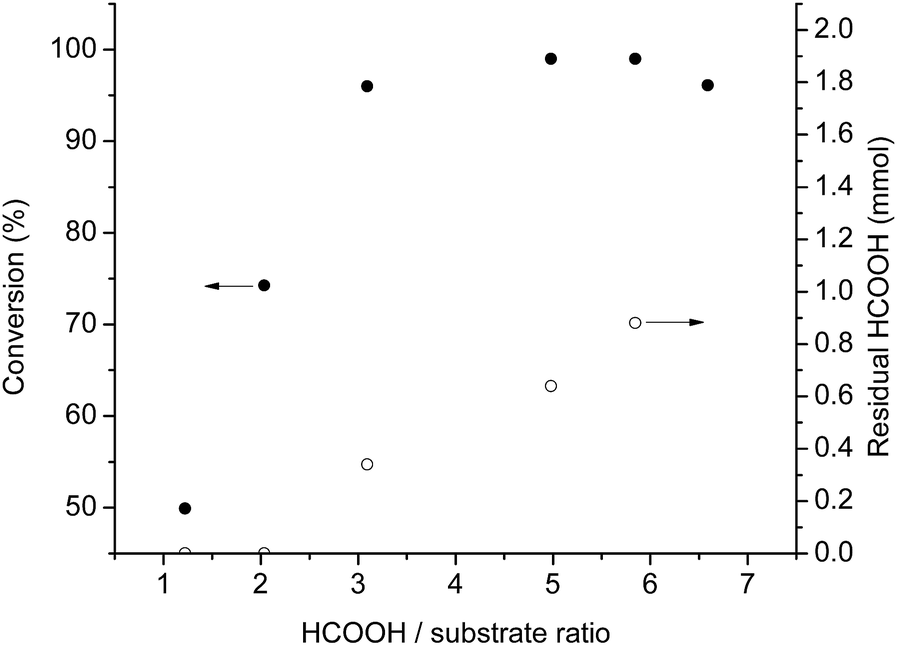 | ||
| Fig. 3 Formation of 1-phenylethanol at different HCOOH/substrate molar ratio. Reaction conditions: 0.005 mmol [Rh(cod)2]+[BF4]−, 0.3 mmol acetophenone, 0.5 mL [TBA][HV], T = 80 °C, t = 3 h. ●: Conversion, ○: residual HCOOH (mmol). | ||
The conversion reached a plateau between 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 6:1 molar ratio (Fig. 3), similarly to the observed case of asymmetric transfer hydrogenation of ketones, using azeotropic mixture of HCOOH/triethylamine as hydrogen donor.29g In extreme cases, such as when the ratio was over 8:1, the conversion decreased significantly (below 20%) presumably due to the dilution and increased acidity of the system, which could result in the loss of the catalyst's activity. The exact amount of HCOOH remained after the completed reaction was determined by 1H-NMR spectroscopy by using GVL as an internal standard in the presence of 0.3 mmol acetophenone, 0.005 mmol [Rh(cod)2]+[BF4]− in 0.5 mL [TBA][HV]. When the HCOOH to substrate ratio was 1:1 and 1.2:1, no residual HCOOH was detected after 3 hours, while in case of 3:1 and 5:1 ratio 40% decrease of the initial amount of H-donor could be obtained. At ratio 6:1 more residual amount of formic acid was detected with full conversion of substrate. Further increasing this ratio resulted a slight decrease of the conversion rate. It is important to note that no water formation was detected. Consequently, due to the decomposition of formic acid, an excess of hydrogen donor has to be used to reach full conversion.
:1 and 6:1 molar ratio (Fig. 3), similarly to the observed case of asymmetric transfer hydrogenation of ketones, using azeotropic mixture of HCOOH/triethylamine as hydrogen donor.29g In extreme cases, such as when the ratio was over 8:1, the conversion decreased significantly (below 20%) presumably due to the dilution and increased acidity of the system, which could result in the loss of the catalyst's activity. The exact amount of HCOOH remained after the completed reaction was determined by 1H-NMR spectroscopy by using GVL as an internal standard in the presence of 0.3 mmol acetophenone, 0.005 mmol [Rh(cod)2]+[BF4]− in 0.5 mL [TBA][HV]. When the HCOOH to substrate ratio was 1:1 and 1.2:1, no residual HCOOH was detected after 3 hours, while in case of 3:1 and 5:1 ratio 40% decrease of the initial amount of H-donor could be obtained. At ratio 6:1 more residual amount of formic acid was detected with full conversion of substrate. Further increasing this ratio resulted a slight decrease of the conversion rate. It is important to note that no water formation was detected. Consequently, due to the decomposition of formic acid, an excess of hydrogen donor has to be used to reach full conversion.
It was proven that the GVL-based ILs can be easily fine-tuned by varying the chain length of the alkyl groups on the cation, resulting in significant changes in viscosity and vapour pressure. In order to determine the influence of the structure on the performance of the catalytic system, we screened a series of GVL-based ILs having different tetraalkylammonium cations. Table 3 shows, that negligible conversion could be achieved using [TMA][HV], which has relatively high melting point (75–80 °C), thus it becomes liquid at the reaction temperature and might be more useful at even higher temperatures. The replacement of methyl to ethyl group resulted in significantly higher conversion (94%, see Table 3, entry 2). Interestingly, further increase of the length of the alkyl chain on the cation had no dramatic effect on the product formation. The reaction rate reached >99% in case of [TBA][HV] (Table 3, entry 4) similarly as in the conventional hydrogenation reactions using GVL-based ILs containing hydroxyvalerate anion.
| Entry | Ionic liquid | Conv. (%) | TOF (h−1) |
|---|---|---|---|
| a Reaction conditions: 0.005 mmol [Rh(cod)2]+[BF4]−, 0.3 mmol acetophenone, 1.6 mmol HCOOH, 0.5 mL ionic liquid, T = 80 °C, t = 3 h. | |||
| 1 | [(CH3)4N]+[HV]−, [TMA][HV] | 6 | 1.2 |
| 2 | [(C2H5)4N]+[HV]−, [TEA][HV] | 93 | 18.7 |
| 3 | [(C3H7)4N]+[HV]−, [TPrA][HV] | 96 | 19.2 |
| 4 | [(C4H9)4N]+[HV]−, [TBA][HV] | >99 | 20.0 |
| 5 | [(C5H11)4N]+[HV]−, [TPeA][HV] | 96 | 19.3 |
| 6 | [(C6H13)4N]+[HV]−, [THA][HV] | 94 | 18.8 |
| 7 | [(C4H9)4N]+[MeOV]−, [TBA][MeOV] | >99 | 20.0 |
| 8 | [(C4H9)4N]+[EtOV]−, [TBA][EtOV] | >99 | 20.0 |
Chakraborti's studies indicated that carbonyl group(s) could participate in the hydrogen bond interaction between the oxygen atom of carbonyl species and a hydrogen atom of corresponding ionic liquid to form an activated intermediate that readily reacts with the other substrate of the reaction.20 This observation was also verified for the addition reactions of α,β-unsaturated carbonyl species.21 To investigate the role of the 4-hydroxyl group of the IL's anion in transfer hydrogenation, the reduction of acetophenone was also performed in tetrabutylammonium 4-methoxyvalerate and 4-ethoxyvalerate, (where the 4-OH group was replaced with 4-OMe and 4-OEt ones). In both cases, full conversion was achieved showing that the hydroxyl group has no role in the substrate activation (Table 3, entries 7 and 8).
It was established that the C2-hydrogen of the imidazolium cation could activate the CO group via hydrogen bond formation, as well.20a,b In order to further demonstrate the possible effect of the IL's structure on the efficiency of the system, butylmethyl imidazolium-based ionic liquids [BMIM]+[X]− (X = Cl−, PF6−, OH−, octylsulphonate) were investigated for comparison. Unexpectedly, no conversion could be detected under the previously optimized conditions. Presumably the proposed interaction could not be established under the applied acidic conditions. Similar observation was reported for the selective reduction of chalcone performed in dialkyl imidazolium type ILs.17a It should be noted that the reduction of the carbonyl group could not be achieved even in tetraalkylphosphonium-based ILs. Analogously, no product formation was detected applying conventional organic solvents such as toluene, methanol, ethanol, propanol, and dimethyl sulfoxide.
To check the activity and stability of the catalytic system, a second portion of acetophenone (0.3 mmol) and HCOOH (1.6 mmol) was added to the mixture of the completed reaction after 3 hours. It was observed that the catalyst remained active and the substrate was completely converted in the consecutive runs, as well. However, a slight decrease in the activity could be detected in the fourth cycle, in which 4 hours was needed to reach the full conversion. Since [TBA][HV] was miscible with the product, it was extracted from the catalyst phase by pentane (2 × 0.5 mL) extraction. Subsequently, HCOOH (1.6 mmol) and the substrate (0.3 mmol) were added and the next cycle was started. This procedure was repeated for three times. Although, full conversion was achieved in cycles 1–3, a slight decrease in the activity was detected in the 4th run (Table 4). Similar remarks were described for Rh-catalyzed transfer hydrogenation of chalcone, where toluene was used for the extraction.17 The isolated yield of 1-phenylethanol was 58%.
| Cycle | Time (h) | Conversion |
|---|---|---|
| a Reaction conditions: 0.01 mmol [Rh(cod)2]+[BF4]−, 0.3 mmol acetophenone, 1.6 mmol HCOOH in 1 mL [TBA][HV], T = 80 °C. | ||
| 1 | 3 | >99 |
| 2 | 3 | >99 |
| 3 | 3 | >99 |
| 4 | 4 | >99 |
As already stated, the electronic properties of the substituents on the carbonyl group had significant effect on the reactivity. The 4-substituted acetophenone derivatives having electron donating substituent such as methyl or methoxy group, were found to be less reactive compared to acetophenone (Table 5, entries 1–3). Consequently, the results indicated that the introduction of electron withdrawing groups led to higher conversion rate (Table 5, entries 4–6), as expected. The 2- and 3-substituted derivatives also showed lower reactivity (Table 5, entries 7 and 8). Identical observations were reported for both conventional and asymmetric transfer hydrogenations.30 It is noteworthy that the reduction of 4-nitroacetophenone led to the formation of 1-(4-nitrophenyl)ethanol. Concerning non-aromatic carbonyl compounds, cyclic ketones were reduced in [TBA][HV] under identical conditions and, as it was predicted, full conversion was achieved (Table 6, entries 1 and 2). Regarding the levulinic acid and its methyl ester, it was revealed that both species can be selectively reduced to gamma-valerolactone (Table 6, entries 3 and 4).
|
|
||||
|---|---|---|---|---|
| Entry | R | Time (h) | Conv. (%) | TOF (h−1) |
| a Reaction conditions: 0.005 mmol [Rh(cod)2]+[BF4]−, 0.3 mmol substrate, 1.6 mmol HCOOH, 0.5 mL [TBA][HV], T = 80 °C. | ||||
| 1 | H | 3 | >99 | 20.0 |
| 2 | 4-CH3 | 3 | 17 | 3.4 |
| 3 | 4-OCH3 | 3 | 27 | 5.4 |
| 4 | 4-NO2 | 1.5 | >99 | 40.0 |
| 5 | 4-F | 2.5 | >99 | 15.0 |
| 6 | 4-Br | 2.5 | >99 | 20.0 |
| 7 | 3-Br | 3 | 94 | 18.8 |
| 8 | 2-Br | 3 | 60 | 12.0 |
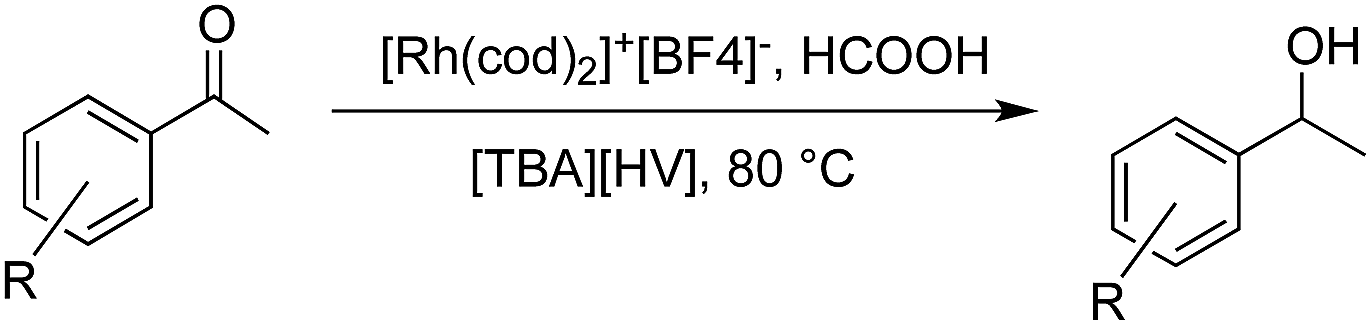
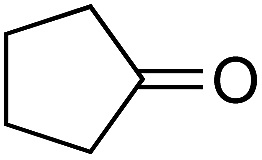
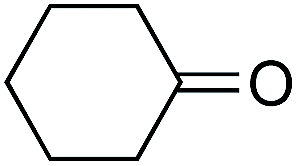
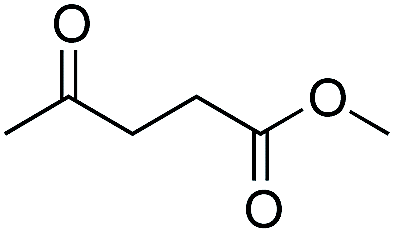
Further investigations were focused on the applicability of [Rh(cod)2]+[BF4]−/HCOOH catalyst system for the hydrogenation of different substrates with olefinic carbon–carbon double bond, under the optimized conditions. It was shown that alk-1-enes could be quantitatively converted to the corresponding alkanes with TOF = 20 h−1 (Table 7, entries 1–3), as expected. For cycloalkenes full conversions were observed (Table 7, entries 4 and 5), similarly to the catalytic hydrogenation of olefins in these ILs using molecular hydrogen. When crotyl bromide and cinnamic acid were subjected to reduction, >99% conversion was achieved for both cases, however, in case of crotyl bromide the reaction was already completed in 1.5 hour (Table 7, entries 6 and 7). For the sterically hindered trans-stilbene slightly lower conversion was observed (Table 7, entry 8).
| Entry | Substrate | Time (h) | Conv. (%) | TOF (h−1) |
|---|---|---|---|---|
| a Reaction conditions: 0.005 mmol [Rh(cod)2]+[BF4]−, 0.3 mmol substrate, 1.6 mmol HCOOH, 0.5 mL [TBA][HV], T = 80 °C. | ||||
| 1 |
|
3 | >99 | 20.0 |
| 2 |
|
3 | >99 | 20.0 |
| 3 |
|
3 | >99 | 20.0 |
| 4 |
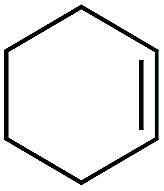
|
3 | >99 | 20.0 |
| 5 |
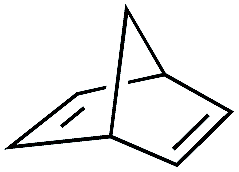
|
3 | >99 | 20.0 |
| 6 |
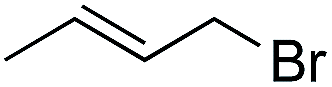
|
1.5 | >99 | 40.0 |
| 7 |

|
3 | >99 | 20.0 |
| 8 |
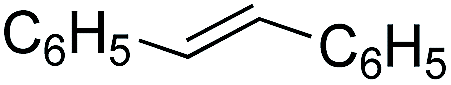
|
3 | 72 | 14.4 |
The selective reduction of α,β-unsaturated carbonyl compounds were investigated by Hermecz et al. in different imidazolium-based ILs using Pd(OAc)2, [Rh(cod)Cl]2 and ClRh(PPh3)3 as catalyst precursors in the presence of HCOONH4 or HCOOH as H-donor.17a When 0.6 mmol of 1-cyclohex-2-enone in the presence of 3.2 mmol HCOOH and 0.01 mmol [Rh(cod)2]+[BF4]− in 1 mL [TBA][HV] was reduced at 80 °C, the complete conversion to cyclohexanone was detected after 15 min; however, after 3 hours both the carbon–carbon double bonds and the carbonyl groups were completely reduced to cyclohexanol. The same results were obtained in case of using chalcone as substrate indicating that the hydrogenation of CC bond is much faster than CO bond. However, by applying longer reaction time both functional groups were reduced. The reduction of cinnamic aldehyde (0.3 mmol) led to the formation of 3-phenylpropanal with conversion of 66% under identical conditions.
The reproducibility of the catalytic reactions were confirmed by repeating the reduction of acetophenone, hex-1-ene, oct-1-ene and dec-1-ene (0.3 mmol) in the presence of HCOOH (1.6 mmol) and [Rh(cod)2]+[BF4]− (0.005 mmol) in [TBA][HV]. Complete conversions were achieved after 3 h at 80 °C in all cases.
Experimental
Tetramethylammonium 4-hydroxyvalerate [TMA][HV], tetrabutylammonium 4-hydroxyvalerate [TBA][HV], tetrahexylammonium 4-hydroxyvalerate [THA][HV], tetrabutyl-ammonium 4-methoxyvalerate [TBA][MeOV] and tetrabutyl-ammonium 4-ethoxyvalerate [TBA][4EtOV] were prepared by published methods.14,20 1-Butyl-3-methylimidazolium hexafluorophosphate, 1-butyl-3-methylimidazolium tetrafluoroborate, 1-butyl-3-methylimidazolium octyl sulfate, chalcone, 1-cyclohex-2-enone, cinnamic acid were purchased from Sigma-Aldrich Kft. and used as received. 1-Butyl-3-methylimidazolium chloride, 1-butyl-3-methylimidazolium hydroxide and 1-ethyl-3-methylimidazolium hydroxide were prepared by published methods.31 The water content of the ILs was determined by Karl-Fischer titration.Preparation of tetraethylammonium 4-hydroxyvalerate [TEA][HV]
The mixture of 14.73 g (100 mmol) of tetraethylammonium 4-hydroxide (in 25% aqueous solution) and 10.51 g (105 mmol) of γ-valerolactone was stirred at room temperature for 1 hour. After the reaction was completed, the residual water was removed in vacuo at 80 °C. The product was isolated as a colourless, highly viscous liquid. Yield 22.11 g (87.6%). 1H-NMR (D2O) δ, ppm: 0.98 (d, 3H, J = 6.5 Hz), 1.07 (t, 12H, J = 7.1 Hz), 1.52 (q, 2H, J = 7.2 Hz), 2.04 (m, 4H), 3.07 (q, 8H, J = 7.1 Hz), 3.62 (m, 1H, J = 6.1 Hz) 13C-NMR (D2O) δ, ppm: 10.51, 25.7, 37.95, 38.72, 55.80 (t, J = 3.3 Hz), 71.5, 186.8 (ESI Fig. S1 and S2†). H2O (wt%): 2.1%.Preparation of tetrapropylammonium 4-hydroxyvalerate [TPrA][HV]
The mixture of 10.2 g (50 mmol) of tetrapropylammonium 4-hydroxide (in 20% aqueous solution) and 5.26 g (53 mmol) of γ-valerolactone was stirred at room temperature for 1 hour. After the reaction was completed, the residual water was removed in vacuo at 80 °C. The product was isolated as a colourless, highly viscous liquid. Yield 15.25 g (98.6%). 1H-NMR (D2O) δ, ppm: 0.81 (t, 12H, J = 7.1 Hz), 1.04 (d, 3H, J = 6.3 Hz), 1.57 (m, 8H), 2.09 (m, 2H), 3.02 (m, 8H), 3.67 (m, 1H, J = 6.3 Hz) 13C-NMR (D2O) δ, ppm: 9.79, 14.76, 21.65, 33.92, 34.69, 59.8 (t, J = 2.6 Hz), 67.57, 183.05 (ESI Fig. S3 and S4†). H2O (wt%): 4.2%.Preparation of tetrapentylammonium 4-hydroxyvalerate [TPeA][HV]
The mixture of 15.8 g (50 mmol) of tetrapentylammonium 4-hydroxide (in 20% aqueous solution) and 5.50 g (55 mmol) of γ-valerolactone was stirred at room temperature for 1 hour. After the reaction was completed, the residual water was removed in vacuo at 80 °C. The product was isolated as a colourless, highly viscous liquid. Yield 19.83 g (93.1%). 1H-NMR (D2O) δ, ppm: 0.80 (t, 12H, J = 6.5 Hz), 1.06 (d, 3H, J = 6.3 Hz), 1.24 (m, 16H), 1.55 (m, 10H), 2.29 (t, 2H, J = 7.1 Hz), 3.08 (m, 8H, J = 7.3 Hz), 3.7 (m, 1H) 13C-NMR (D2O) δ, ppm: 13.17, 20.81, 21.58, 21.80, 27.79, 34.08, 34.82, 58.26 (t, J = 3.60 Hz), 67.71, 183.03 ESI (Fig. S5 and S6†). H2O (wt%): 2.6%.The vapour pressure of the ionic liquids were measured by using a Rosemount manometer attached to a 25 mL Hastelloy-C high-pressure reactor (Parr Inst., IL, USA) equipped with magnetic stirrer and a thermocouple. The latter was connected to the computer through an AD interface to register the actual temperature. Initially, the reactor was charged with 1 mL of the corresponding ionic liquid, followed by the attachment of the manometer, and de-aerated at room temperature. The reactor was then placed into ice–water mixture. The exact value of the vapour pressure was recorded when it was stabilized at the desired temperature.
The transfer hydrogenation reactions were performed in Hach tubes (100 × 13 mm) closed with screw-cap. In a typical experiment, 0.005 mmol of the correspondent catalyst was dissolved in 0.5 mL ionic liquid, then 1.5 mmol H-donor and 0.3 mmol substrate was added to the solution. The reaction mixture was stirred at 80 °C for 3 h. The conversion and yield were determined by GC analysis and 1H-NMR spectroscopy.
In order to determine the amount of residual HCOOH in the reaction, equimolar amount of HCOOH and GVL (as internal standard) was added to the mixture of 0.5 mL [TBA][HV] and 0.3 mmol acetophenone. A sample was taken before heating up the mixture and another one after the reaction was completed. The samples were dissolved in d6-acetone, then analyzed by 1H-NMR.
The NMR spectra were recorded on a Brucker Avance 250 spectrometer.
In the recyclability experiments 0.3 mmol of acetophenone was reduced by using 0.01 mmol of [Rh(cod)2]+[BF4]− in 1 mL [TBA][HV] at 80 °C. After completing the reaction the product was extracted with 2 × 0.5 mL pentane. To initiate the following cycle, acetophenone (0.3 mmol) and HCOOH (1.6 mmol) were added to the catalyst phase followed by heating up to 80 °C. An intensive gas evolution indicated the start of the reaction. The procedure was repeated 3 times and the isolated products from the consecutive cycles were analyzed by GC.
GC analyses were performed on an HP 5890N instrument with a HP-5 capillary column (15 m × 0.25 μm) using H2 as a carrier gas. For the analysis, 5 μL of reaction mixture was dissolved in 1 mL of methylene chloride followed by the addition of 5 μL toluene as internal standard.
Conclusions
γ-Valerolactone-based ionic liquids (ILs) containing hydroxyvalerate anion were successfully applied as alternative solvents for homogenous catalytic transfer hydrogenation of acetophenone and its substituted forms, functionalized ketones and alkenes. The highest conversions were achieved by using [Rh(cod)2]+[BF4]− as catalyst precursor and formic acid as hydrogen donor at 80 °C, with the molar ratio of HCOOH/substrate between 5:1 and 6:1. A series of GVL-based ILs with tetraalkylammonium cations were tested showing negligible influence of the cation structure on the catalytic activity. The potential recyclability of the catalytic system was demonstrated in four consecutive cycles for the reduction of acetophenone.
Acknowledgements
This work was funded by the Budapest University of Technology and Economics under project number KMR_12-1-2012-0066 and the Bolyai János Research Scholarship of the Hungarian Academy of Sciences.Notes and references
-
(a)
F. M. Kerton, Alternative Solvents for Green Chemistry, RSC, Cambridge, 2009 Search PubMed
; (b) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, UK, 1998 Search PubMed
-
(a)
Water in Organic Synthesis, ed. S. Kobayashi, Georg Thieme Verlag KG, Stuttgart, 2012 Search PubMed
-
(a)
Handbook of Fluorous Chemistry, ed. J. A. Gladysz, D. P. Curran and I. T. Horváth, Wiley-VCH, Weinheim, 2005 Search PubMed
-
Handbook of Green Chemistry, in Supercritical Solvents, ed. W. Leitner and P. G. Jessop, Wiley-VCH, Weinheim, vol. 4, 2010 Search PubMed
- W. Keim, Chem. Ing. Tech., 1984, 56, 850 CrossRef CAS
-
D. J. Adams, P. J. Dyson and S. J. Taverner, Chemistry in Alternative Reaction Media, John Wiley & Sons Ltd, Chichester, 2004 Search PubMed
-
(a) H. Olivier-Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1 CrossRef CAS
-
(a) T. Welton, Chem. Rev., 2009, 99, 2071 CrossRef
-
(a) S. Csihony, H. Mehdi and I. T. Horváth, Green Chem., 2001, 3, 307 RSC
- C. P. Mehnert, R. A. Coock, N. C. Dispenziere and E. J. Mozeleski, Polyhedron, 2004, 23, 2679 CrossRef CAS
- V. Fábos, D. Lantos, A. Bodor, A. M. Bálint, L. T. Mika, O. E. Sielcken, A. Cuiper and I. T. Horváth, ChemSusChem, 2008, 1, 189 CrossRef PubMed
-
(a)
P. A. Chaloner, M. A. Esteruleas, F. Joó and L. Oro, Homogeneous Hydrogenation, Kulwer Academic Press, Dordrect, 1994 Search PubMed
-
(a) Y. Chauvin, L. Mussmann and H. Olivier, Angew. Chem., Int. Ed. Engl., 1995, 34, 2698 CrossRef CAS
- A. Strádi, M. Molnár, M. Óvári, G. Dibó, F. U. Richter and L. T. Mika, Green Chem., 2013, 15, 1857 RSC
-
(a) G. Brieger and T. J. Nestrick, Chem. Rev., 1974, 74, 567 CrossRef CAS
- H. Berthold, T. Schotten and H. Hönig, Synthesis, 2002, 11, 1607 Search PubMed
-
(a) Z. Baán, Z. Finta, G. Keglevich and I. Hermecz, Green Chem., 2009, 11, 1937 RSC
- I. Kawasaki, K. Tsunoda, T. Tsuji, T. Yamaguchi, H. Shibuta, N. Uchida, M. Yamashita and S. Ohta, Chem. Commun., 2005, 2134 RSC
- T. J. Geldbach and P. Dyson, J. Am. Chem. Soc., 2004, 126, 8114 CrossRef CAS PubMed
-
(a) A. K. Chakraborti and S. R. Roy, J. Am. Chem. Soc., 2009, 131, 6902 CrossRef CAS PubMed
-
(a) A. Sarkar, S. A. Roy and A. K. Chakraborti, Chem. Commun., 2011, 47, 4538 RSC
- D. Fegyverneki, L. Orha, G. Láng and I. T. Horváth, Tetrahedron, 2010, 66, 1078 CrossRef CAS
- I. T. Horváth, H. Mehdi, V. Fábos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238 RSC
- C. Comyns, N. Karodia, S. Zeler and J. A. Andersen, Catal. Lett., 2000, 67, 113 CrossRef CAS
- V. Fábos, L. T. Mika and I. T. Horváth, Organometallics, 2014, 33, 181 CrossRef
- J. M. Brunel, Tetrahedron, 2007, 63, 3899 CrossRef CAS
- R. Xing, W. Qi and G. W. Huber, Energy Environ. Sci., 2011, 4, 2193 CAS
- A. Bényei and F. Joó, J. Mol. Catal., 1990, 58, 151 CrossRef
-
(a) C. Wang, X. F. Wu and J. L. Xiao, Chem.–Asian J., 2008, 3, 1750 CrossRef CAS PubMed
-
(a) M. Aydemir, N. Meric, C. Kayan, F. Ok and A. Baysal, Inorg. Chim. Acta, 2013, 398, 1 CrossRef CAS
-
(a) P. J. Dyson, M. C. Grossel, N. Srinivasan, T. Vine, T. Welton, D. J. Williams, A. J. P. White and T. Zigras, J. Chem. Soc., Dalton Trans., 1997, 3465 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra08297h |
| This journal is © The Royal Society of Chemistry 2015 |