
Nickel doped nanohydroxyapatite: vascular endothelial growth factor inducing biomaterial for bone tissue engineering†
B. Anu Priyaa,
K. Senthilgurua,
T. Agarwala,
S. N. Gautham Hari Narayanaa,
S. Girib,
K. Pramanika,
K. Pala and
I. Banerjee*a
aDepartment of Biotechnology & Medical Engineering, National Institute of Technology, Rourkela-769008, Odisha, India. E-mail: banerjeei@nitrkl.ac.in; Tel: +91-943-850-7035
bDepartment of Chemistry, National Institute of Technology, Rourkela-769008, Odisha, India
First published on 17th August 2015
Abstract
Biomaterial induced activation of vascular endothelial growth factor (VEGF) pathway for angiogenesis is now gaining recognition as an effective option for tissue engineering. In this context, bivalent nickel (Ni2+) ion doped nano-hydroxyapatites (nHAp) were synthesized by wet chemical method, characterized and evaluated for their osteoconductive and proangiogenic properties. Electron microscopy (FESEM and TEM) along with ICP-OES analysis ensured formation of Ni2+ doped nanoparticles (average ferret diameter 15–17 nm). The ‘apatite identity’ of the nanoparticles was confirmed by XRD and FT-IR. Analysis revealed that Ni2+ doping caused no significant distortion in the crystal structure but decreased the crystallinity of nHAp considerably. Similarly, no major variation in the surface area (BET analysis), the zeta potential and the protein adsorption was observed among the samples. Biological characterization showed that Ni2+ doping influenced the cell viability, proliferation and differentiation of bone cells (MG-63) in a concentration dependent way. ELISA and RT-PCR study revealed that Ni2+ doped nHAp induced cellular VEGF expression many fold in comparison to control. Profiling of hypoxia inducible factor 1 alpha (HIF-1α) expression by immuno-cytochemistry and RT-PCR implied its involvement in cellular VEGF production. In conclusion, Ni2+ doped nHAp may serve as proangiogenic–osteogenic biomaterial for bone tissue engineering.
1. Introduction
One of the major challenges associated with the clinical translation of scaffold-based bone tissue engineering is to ensure vascularization after implantation in vivo. Vascularization is essential for the transport of the nutrients and gases to the cells present at the distal location of an implanted scaffold which can hardly be reached through interstitial fluid diffusion (diffusion transport limit is 200 μm approx.).1,2 An inadequate vascularization often hinders the formation of the regenerative callus resulting in atrophic non-union of the bone. During last couple of years, numerous strategies have been implemented to promote vascularization of bone tissue constructs. These include the use of angiogenic growth factors (VEGF, PDGF) to induce endothelial cell proliferation and migration,3,4 direct application of endothelial or its progenitor cell,5 development of microfluidic based network inside the scaffold (analogues to natural vascularization),6 and the activation of the genetic-network of the cells for enhanced production of angiogenic growth factors. The short half-life, high cost and uncontrolled 3D distribution in vivo limits the application of angiogenic growth factors in the bone tissue engineering. The cell-based approach of promoting angiogenesis failed to meet the expectation for a number of reasons. Firstly, a great phenotypic variation exists among endothelial cells (depending on the source, organ and tissue location) which profoundly influence the angiogenic potential, molecular permeability, homeostasis, vascular tone and even immune tolerance. Secondly, a successful synthesis of vasculature needs a controlled co-culture of a number of cells along with the endothelial cells, which is difficult to achieve.7 The strategies pertaining to the development of microfluidic network based vasculature analogue involves sophisticated and costly infrastructure which limit its translation. Among the aforesaid vascularization strategies, gene therapy is probably the most cost-effective method. However, the difficulty lies in targeting specific host cells and the risk of introduction of vector gene into the patient.8,9Recently, research has been focussed on biomaterials that are capable of stimulating the cells for biased production of angiogenic factors in vivo. In this regard, a common approach is to create a tissue hypoxia condition in vivo. It is already known that angiogenesis get induced by hypoxic condition through HIF-1α (hypoxia inducible factor-1α) pathway. Moreover, hypoxia couples angiogenesis to osteogenesis.10 HIF-1α is a transcription factor, responsible for activation of VEGF gene which in turn produces VEGF, a potential mitogen for endothelial cells. Under normoxic condition, prolyl hydroxylase (PH) induces ubiquitin mediated proteosomal degradation of HIF-1α.11 In the absence of oxygen, PH fails to degrade HIF-1α leading to an increase in VEGF production. It is important to mention that metal ions including cobalt (Co2+), copper (Cu2+), chromium (Cr3+), titanium (Ti2+), and vanadium (V3+) stabilize the HIF-1α by inactivating the PH enzyme.12–16 This results in the accumulation of intracellular HIF-1α (a biochemical milieu, analogues to the hypoxic condition) and subsequently increases the production of VEGF. Exploiting the aforesaid strategies, Wu et al. (2012 and 2013) showed that copper and cobalt doped mesoporous bioactive glass scaffolds can effectively induce cellular VEGF production and angiogenesis.17,18
Nickel is a group ‘d’ metal. The average quantity of nickel present in healthy individual is about 10 mg (0.1 ppm).19 The concentration of nickel present in the serum is approximately 0.2 μg l−1 in normal population.19 Earlier nickel was considered harmful to human health, causing skin allergies, asthma, inflammation, lung fibrosis, kidney diseases and cancer.20 Lately, it was realized that the nickel toxicity is dose-dependent therefore, can be used safely in vivo with certain modifications/precautions. Owing to these facts, nickel based materials like stainless steel (15% Ni) and a number of shape-memory alloys like nitinol (50% Ni)21,22 have now been used in biomedical implants for clinical application. One interesting property of nickel (especially of Ni2+) is that like many other ‘d’ block elements (Cu, Co, Cr, Ti, V), it can also stabilize the HIF-1α, therefore, can induce cellular VEGF secretion by creating intracellular hypoxia mimicking condition.23 Keeping the aforesaid perspective in mind, we hypothesized that incorporation of Ni2+ in an osteogenic biomaterial may convert it into a proangiogenic–osteogenic biomaterial which could be a better option for bone tissue engineering.
Synthetic nano-hydroxyapatite (nHAp) has already been regarded as one of the best biomaterials for bone tissue engineering because of its structural and chemical resemblance with bone apatite and for its osteoconductive properties. In natural bone, nano-sized needle-like hydroxyapatite crystals (HAp) are grown in intimate contact with collagen fibers. One interesting feature of the normal bone apatite is that they are calcium deficient in nature and can efficiently exchange cations (Ca2+) and anions [OH− and (PO4)−3] with the surroundings. It was realized later that the substitution of such ions play a predominant role in the biochemical aspects of these hard tissues. Therefore, replacement of the calcium and hydroxide ions in the synthetic nHAp crystal structure with various metal ions (e.g. Mg2+, Zn2+, Mn2+, Co2+, Y3+) has been done for various applications including bone tissue engineering.24 In relation to our hypothesis, this manuscript delineates the synthesis, physicochemical characterization and biological performance (osteogenic and angiogenic properties) of Ni2+ doped nHAp. Very few reports are available on the synthesis, characterization and application of Ni2+ doped HAp.25,26 However, none of the studies pertain to the exploration of proangiogenic–osteogenic properties of Ni2+ doped HAp.
Here, Ni2+ doped nHAp samples were synthesized via wet chemical precipitation method. The physico-chemical characterization of the synthesized nHAp was carried out by XRD, FT-IR, FESEM, TEM, Zeta analyser, BET surface analyser and protein adsorption profiling. Biological properties including osteogenic and proangiogenic potentials of doped nHAp were assessed in vitro by cell cycle analysis, live-dead assay, MTT assay, immunocytochemistry, ELISA and RT-PCR using human osteoblast cell lines (MG-63).
2. Materials and methods
2.1. Materials
Calcium nitrate tetrahydrate (Ca(NO3)2·4H2O), diammonium hydrogen phosphate ((NH4)2HPO4), nickel nitrate hexahydrate (Ni(NO3)2·6H2O), cetyltrimethylammonium bromide (CTAB) and ammonia solution (25%) were purchased from Merck, India. Dulbecco's Modified Eagle Media (DMEM), Dulbecco's Phosphate Buffer Saline (DPBS) solution, trypsin-EDTA solution, fetal bovine serum, antibiotic–antimycotic solution, and MTT assay kit were purchased from Himedia, India. Human VEGF ELISA kit was purchased from Abcam, UK. mRNA isolation kit (RNeasy Mini) and cDNA synthesis kit (MuLV Reverse Transcriptase Plus Kit) were bought from Qiagen and BioBharti LifeSciences Pvt. Ltd, India, respectively. The MG-63 cell line was procured from NCCS, India.2.2. Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w) was added drop-wise to the Ca(NO3)2·4H2O solution at a constant rate of 1.5 ml min−1 from a burette. The slurry was allowed to age for 24 h. The slurry obtained was then washed by centrifuging at 6000 rpm for 10 min to remove the unreacted reactants. The precipitate was then dried at 55 °C for 24 h to get the nHAp powder (control – designated as HAP). Nickel doped nHAp samples were prepared by the same method in which different percentage of (Ni(NO3)2·6H2O) (1%, 5%, and 10% w/w) was first added to the Ca(NO3)2·4H2O solution. The molar ratio of Ca/P and (Ca + Ni)/P in all the cases were maintained at 1.67. The dried samples were then ball-milled (Pulverlsette 5 planetary ball mill, Fritsch) for 6 h at 350 rpm. nHAp samples corresponding to 1%, 5% and 10% Ni2+ doping were designated as N1-HAP, N5-HAP and N10-HAp, respectively.
:1 (w/w) was added drop-wise to the Ca(NO3)2·4H2O solution at a constant rate of 1.5 ml min−1 from a burette. The slurry was allowed to age for 24 h. The slurry obtained was then washed by centrifuging at 6000 rpm for 10 min to remove the unreacted reactants. The precipitate was then dried at 55 °C for 24 h to get the nHAp powder (control – designated as HAP). Nickel doped nHAp samples were prepared by the same method in which different percentage of (Ni(NO3)2·6H2O) (1%, 5%, and 10% w/w) was first added to the Ca(NO3)2·4H2O solution. The molar ratio of Ca/P and (Ca + Ni)/P in all the cases were maintained at 1.67. The dried samples were then ball-milled (Pulverlsette 5 planetary ball mill, Fritsch) for 6 h at 350 rpm. nHAp samples corresponding to 1%, 5% and 10% Ni2+ doping were designated as N1-HAP, N5-HAP and N10-HAp, respectively.| KA = B1/2 × (Xc)1/3 |
Further, the lattice parameters ‘a’ and ‘c’ of HAp crystal (hexagonal system) were calculated using the formula given below
| a = (6/√3) × d(300) |
| c = 2 × d(002) |
| 1/dhkl = [4 (h2 + k2 + hk)/3a2 + l2/c2]1/2 |
FT-IR analysis of the samples was done by FT-IR spectrophotometer (IR prestige 21, Shimadzu) using KBr pellet. Scanning was done over the range of 400 cm−1–4000 cm−1 in transmittance mode.30 Extent of doping was measured by inductively coupled plasma optical emission spectrometry (ICP-OES) (Perkin-Elmer optima 5300 DV).31 Sample preparation for ICP-OES was done as per the protocol described by Wadekar et al. (2006).32 In brief, the samples were digested with mixture of conc. nitric acid (69%) and conc. perchloric acid (70%) in ratio of 2:1 (% v/v). The concentrated acid digest was heated at 50 °C continuously on a hot plate to remove concentrated acids and the solution was diluted with 1% nitric acid. Final volume of the digested samples was made upto 25 ml with distilled water and the pH of the final solution was adjusted to ∼6–7 for ICP-OES analysis. To study the morphology and particle size, both scanning electron and transmission electron microscopy were done. For scanning electron microscopy, powdered samples were sputter coated with gold and then visualized under FESEM (FEI, Nova NanoSEM) at 3 kV.33 For transmission electron microscopy, a drop of dilute suspension of the powdered samples were placed on 100 μm mesh carbon-coated copper grid and dried overnight under vacuum at room temperature. Samples were then investigated using High Resolution Transmission Electron Microscope (JEM 2100, JOEL) at 200 kV.34 BET surface area analyser (Autosorb-1, Quantachrome Instruments) was used to determine the surface area, pore size and pore volume of the doped samples. The analysis was done following 5-point fast scan procedure.34 The zeta potential of the nHAp samples was determined using zeta analyser (Nano ZS, Marvelin). For this analysis, 1 mg of the sample was added in 10 ml of injection water (pH 7.0) and sonicated for 10–15 min to break the agglomeration. Then, the suspension was immediately subjected to zeta analysis.35 For protein adsorption study, 10 mg of nHAp powder was incubated in 1 ml of an aqueous solution of bovine serum albumin (BSA) (1200 μg ml−1) at 37 °C for 24 h. After incubation, the solution was centrifuged at 8000 rpm for 10 min and the supernatant was collected. The residual protein concentration in the supernatant was determined using Bradford assay. The adsorption of the proteins by the samples were determined by subtracting the residual protein concentration from the total protein concentration.36
The expression of the differentiation marker Runx2 (Runt-related transcription factor 2) and HIF-1α was analyzed using immunocytochemistry. For the analysis, cells were treated with the samples at a concentration of 100 μg ml−1. The primary antibodies used for the analysis were anti-HIF-1α antibody (mouse, anti-human, 1:200 dilution) and anti-Runx2 antibody (rabbit anti-human, 1:200 dilution). Secondary antibodies were anti-Rabbit Alexa Flour 488 and anti-Mouse Alexa Flour 488. Actin cytoskeleton and nuclear staining was done using TRITC-phalloidin and Hoechst dyes. The samples were visualized under the confocal microscope (TCS-SP8, Leica), keeping all the imaging parameters same.40 The image quantification was carried out using NIH ImageJ software. VEGF expression from the MG-63 cells was determined using human VEGF ELISA kit (Abcam 100662) as per the manufacturer's instructions.41 For the analysis, cells were treated with the samples at a concentration of 100 μg ml−1 for 48 h and the spent media was used to quantify secreted VEGF. Furthermore, expression of VEGF, Runx2, and HIF-1α in MG-63 was also checked by RT-PCR. For this, the cells were treated with the samples at a concentration of 100 μg ml−1 for 48 h. mRNA isolation and cDNA synthesis was carried out using Qiagen RNeasy Mini Kit and BioBharti MuLV Reverse Transcriptase plus Kit, respectively, following the manufacturer's instructions. Further, the gene specific amplification was done using PCR and expression profile was assessed by comparing band intensity. GAPDH was used as a control for the analysis. For the amplification, the primers used include 5′ GCACCCATGGCAGAAGGAG 3′ (VEGF_F), 5′ ACACAGGATGGCTTGAAGATGT 3′ (VEGF_R), 5′ GTTTACTAAAGGACAAGTCACC 3′ (HIF1_F), 5′ TTCTGTTTGTTGAAGGGAG 3′ (HIF1_R), 5′ TCTGGCCTTCCACTCTCAGT 3′ (Runx2_F), 5′ GACTGGCGGGGTGTAAGTAA 3′ (Runx2_R), 5′ CATGAGAAGTATGACAACAGCCT 3′ (GAPDH_F) and 5′ AGTCCTTCCACGATACCAAAGT 3′ (GAPDH_R).
3. Results
3.1. Preparation of Ni2+ doped nHAp
Many research groups has prepared nHAp and doped nHAp using number of methodologies such as hydrothermal synthesis, emulsion method, solid state synthesis, wet chemical method, sol–gel method and biosynthesis.42 Among the techniques mentioned above, wet chemical method has been adopted widely because of process simplicity and controllability. Here, synthesis of nHAp and Ni2+ doped nHAp was done by ammoniacal precipitation method. Cationic surfactant CTAB was used as a capping agent to control the size of the nanoparticle.43 Three different sets of the Ni2+ doped nHAp samples were prepared by varying the doping concentration (Table 1). The yield of nHAp and Ni2+ doped nHAp was between 0.95 to 1.0 g in all the cases. There was no significant variation in the yield with an increase in the doping percentage. For all the cases, percentage yield (w/w) was approximately 30%. In order to gain powder nHAp, the agglomerate was ball-milled after drying. Physical inspection of the powdered nHAp revealed that all the samples were free-flowing. HAP was milky white in colour, while Ni2+ doped nHAp were whitish green (Fig. S1. ESI section†).| S. no | Samples | Composition | [Ca(NO3)2·4H2O + Ni(NO3)2·6H2O] g/200 ml | [(NH4)2HPO4] g/200 ml | Yield (g) | Colour |
|---|---|---|---|---|---|---|
| 1 | HAP | Hydroxyapatite [Ca/P: 1.67] | 2.36 + 0.0 | 0.79 | 1.00 ± 0.03 | Milk white |
| 2 | N1-HAP | Hydroxyapatite + 1% (w/w) nickel [(Ca + Ni)/P: 1.67] | 2.36 + 0.0236 | 0.796 | 0.97 ± 0.01 | Creamy white |
| 3 | N5-HAP | Hydroxyapatite + 5% (w/w) nickel [(Ca + Ni)/P: 1.67] | 2.36 + 0.118 | 0.822 | 0.99 ± 0.09 | Greenish white |
| 4 | N10-HAP | Hydroxyapatite + 10% (w/w) nickel [(Ca + Ni)/P: 1.67] | 2.36 + 0.236 | 0.854 | 1.00 ± 0.01 | Greenish white |
3.2. Physico-chemical characterization
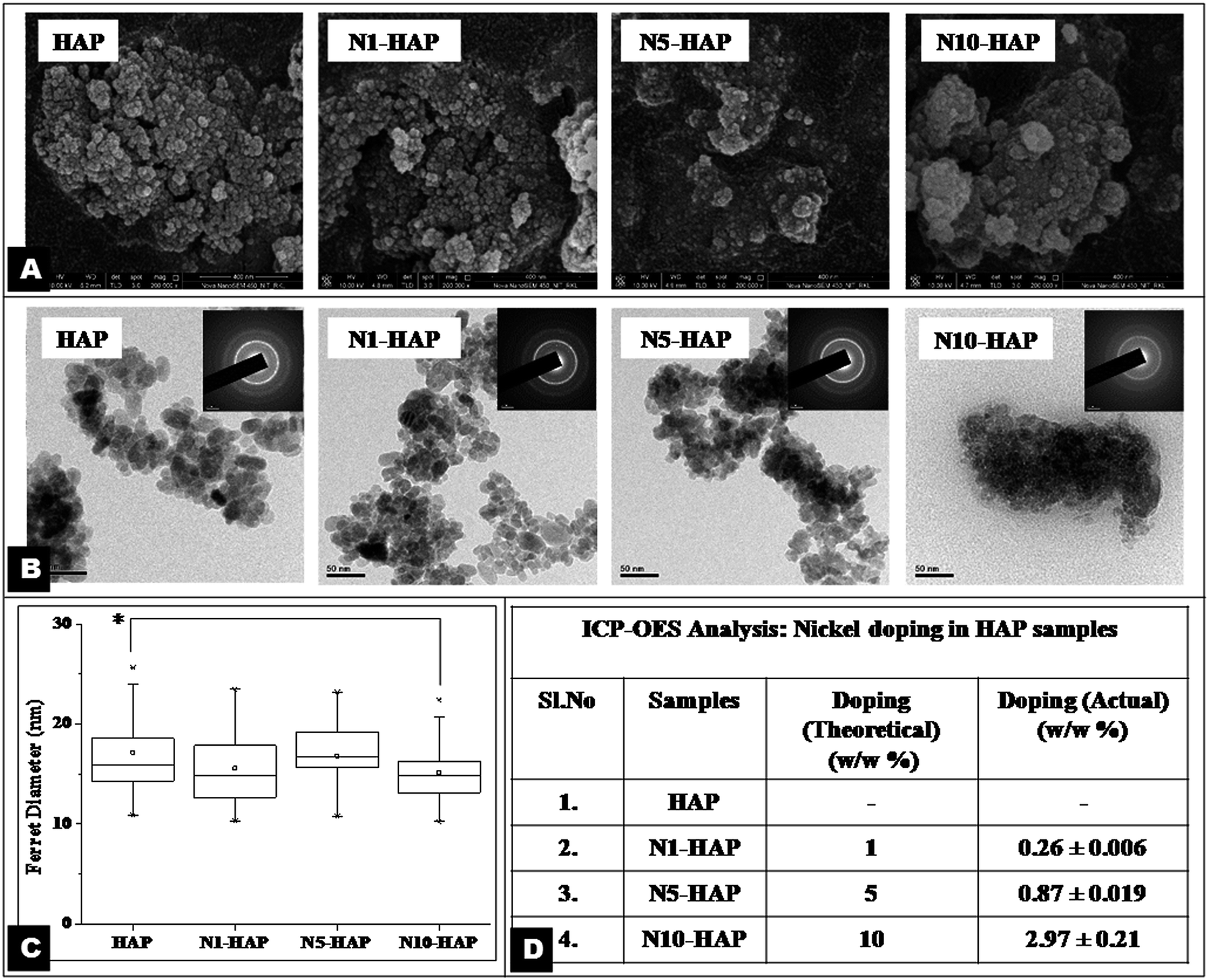 | ||
| Fig. 1 [A] FESEM micrographs of nHAp and Ni2+ doped nHAp. [B] TEM images of nHAp and Ni2+ doped nHAp. [C] Nano particle size distribution. TEM image based analysis was done using NIH-ImageJ software. For each analysis 25 individual particles were considered (p < 0.05). [D] ICP-OES analysis of percentage of Ni2+ doping with respect to calcium content (w/w). Results were expressed as mean ± S.D of three independent experiments (p < 0.05). | ||
| S. no | Samples | % Crystallinity | Crystal lattice parameters (Å) | ‘d’-spacing (nm) | |
|---|---|---|---|---|---|
| ‘a’ | ‘c’ | ||||
| 1 | HAP | 42.0 | 9.37 | 6.88 | 0.3644 |
| 2 | N1-HAP | 42.0 | 9.43 | 6.90 | 0.3642 |
| 3 | N5-HAP | 64.0 | 9.37 | 6.88 | 0.3624 |
| 4 | N10-HAP | 22.0 | 9.43 | 6.90 | 0.3578 |
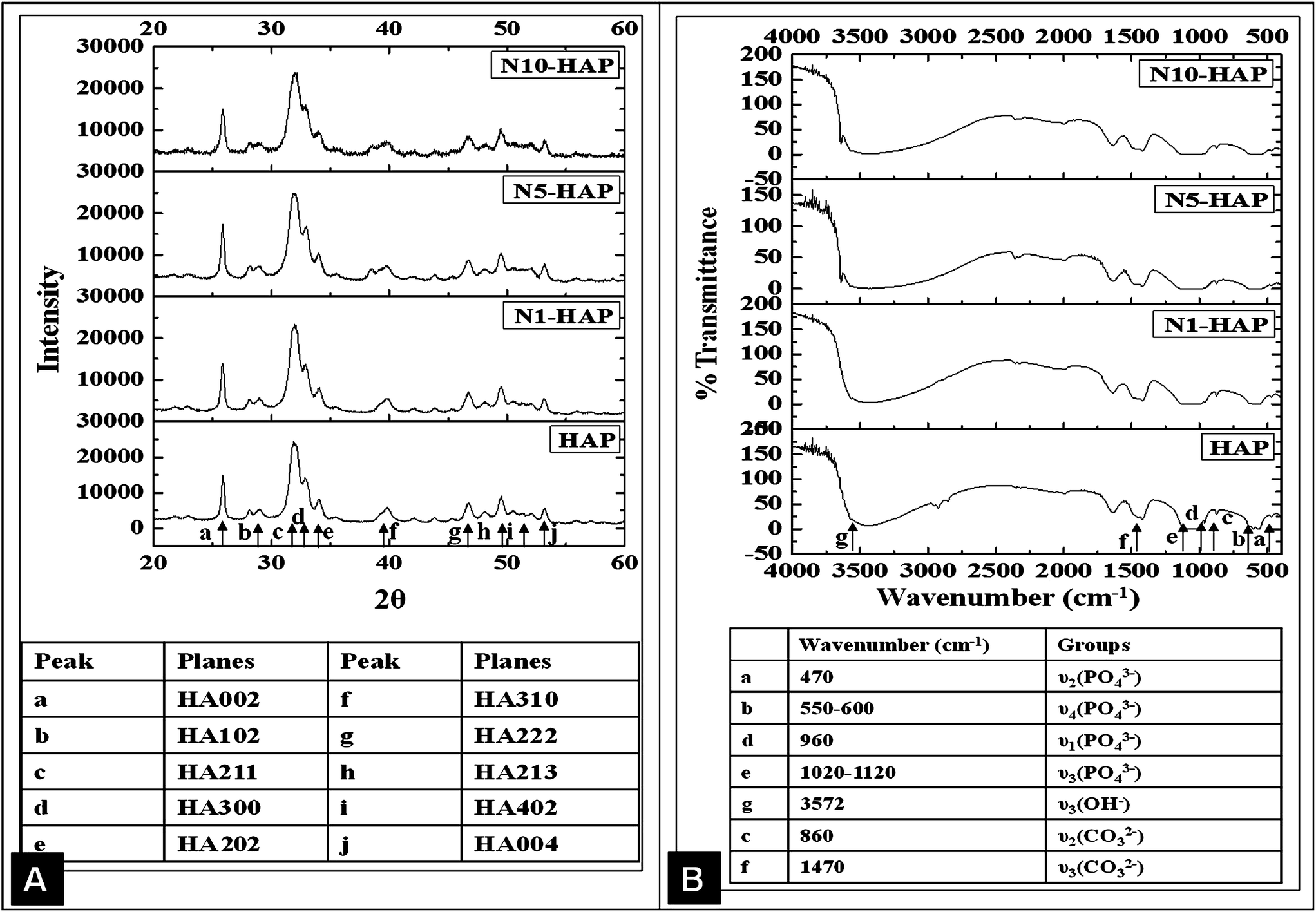 | ||
| Fig. 2 [A] XRD analysis of nHAp and Ni2+ doped nHAp. Important characteristic peaks are marked in the figure, and its corresponding crystal planes were mentioned in the embedded table. [B] FT-IR analysis of nHAp and Ni2+ doped nHAp. Important characteristic peaks are marked in the figure, and its corresponding bond perturbation were mentioned in the embedded table. | ||
| S. no | Samples | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Zeta potential (mV) | Protein adsorption (μg) |
|---|---|---|---|---|---|
| 1 | HAP | 109.1 | 0.041 | −3.5 | 881 ± 19.5 |
| 2 | N1-HAP | 107.6 | 0.040 | −1.0 | 885 ± 7.4 |
| 3 | N5-HAP | 111.7 | 0.046 | 3.2 | 873 ± 18.4 |
| 4 | N10-HAP | 83.2 | 0.034 | 1.1 | 878.5 ± 1.2 |
3.3. Biological characterization
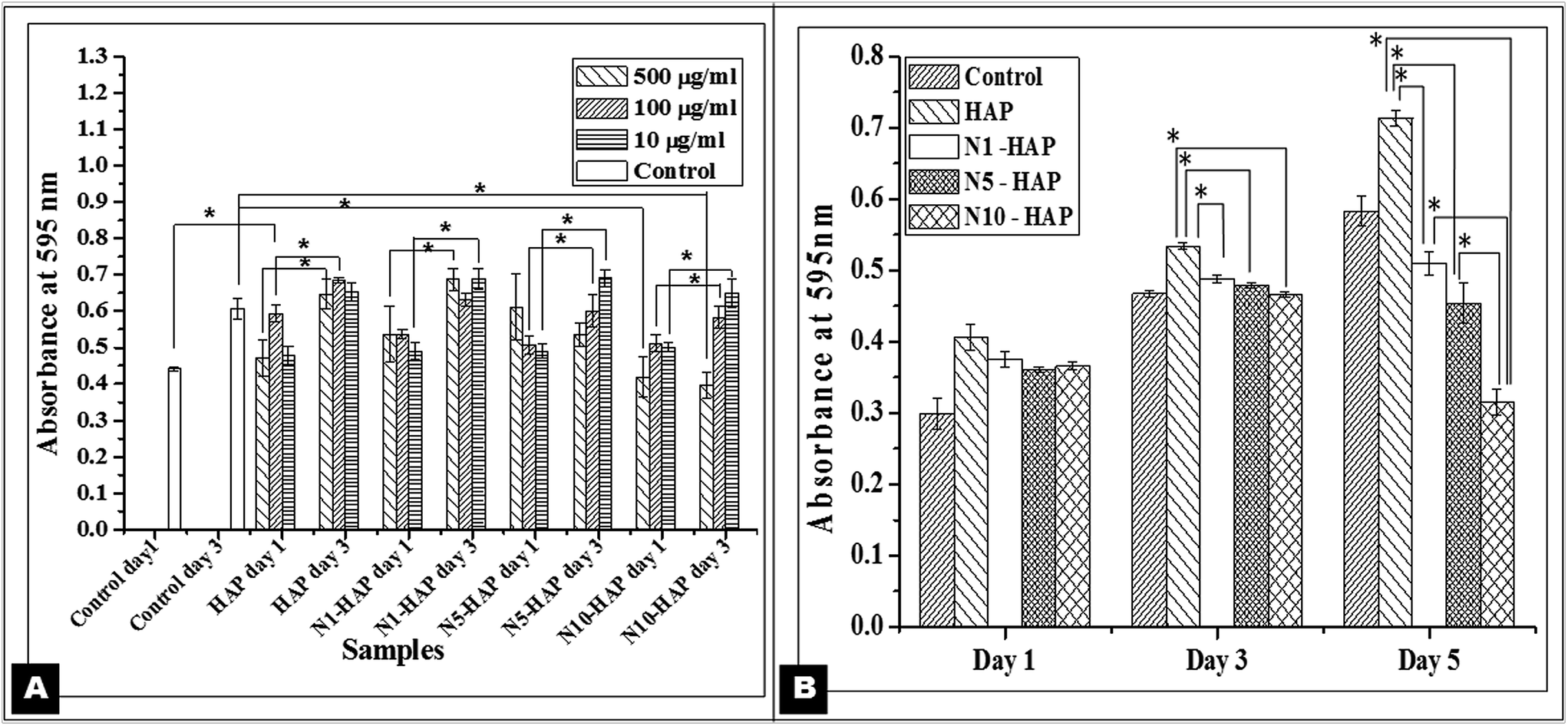 | ||
| Fig. 3 Cell viability and cell proliferation study by MTT assay. [A] Cells were treated with Ni2+ doped nHAp at concentrations of 500 μg ml−1, 100 μg ml−1 and 10 μg ml−1. Data were expressed as mean ± S.D (n = 3). Statistical significance was checked for p < 0.05. [B] Cells were treated with Ni2+ doped nHAp at a concentration of 100 μg ml−1 for the respective days (1, 3 & 5). Data were expressed as mean ± S.D (n = 3). Statistical significance was checked for p < 0.005. | ||
Effect of doped samples (at a concentration of 100 μg ml−1) on long term cell proliferation was further checked by MTT assay (Fig. 3B). HAP showed the highest cell proliferation in comparison to all other samples upto day 5. On day 1, there was no significant variation in the cell proliferation among all the groups (p > 0.005). On day 3, HAP treated cells showed significant proliferation with respect to Ni2+ doped nHAp treated groups (p < 0.005). However, no significant variation in the cell proliferation was observed among N1-HAP, N5-HAP, and N10-HAP. On day 5, significant variation in cell proliferation was observed among all the groups (p < 0.005) where N10-HAP treated cells showed least proliferation. Comparative analysis of the proliferation rate showed that HAP treated cells maintained a consistent growth rate (1.31 fold and 1.33 fold increase in OD value from day 1 to day 3 and day 3 to day 5) which was found highest for all the samples. For all the Ni2+ doped nHAp samples, cell growth from day 1 to day 3 was almost same (1.27 to 1.32 fold increase). But from day 3 to day 5, it was observed that the growth rate was retarded with the increase in the Ni2+ doping percentage in case of N5-HAP (0.94 fold) and N10-HAP (0.67 fold).
| Live dead assay: percentage of live cells | |||||
|---|---|---|---|---|---|
| Control | HAP | N1-HAP | N5-HAP | N10-HAP | |
| Day 1 | 94.9 ± 1.2 | 94.9 ± 1.0 | 93.4 ± 1.5 | 92.7 ± 1.5 | 89.9 ± 2.5 |
| Day 3 | 94.2 ± 0.9 | 91.7 ± 0.5 | 93.3 ± 1.1 | 92.9 ± 0.07 | 94.5 ± 1.1 |
| Day 5 | 95.1 ± 1.8 | 92.1 ± 1.5 | 92.1 ± 1.1 | 91.1 ± 0.9 | 92.3 ± 0.1 |
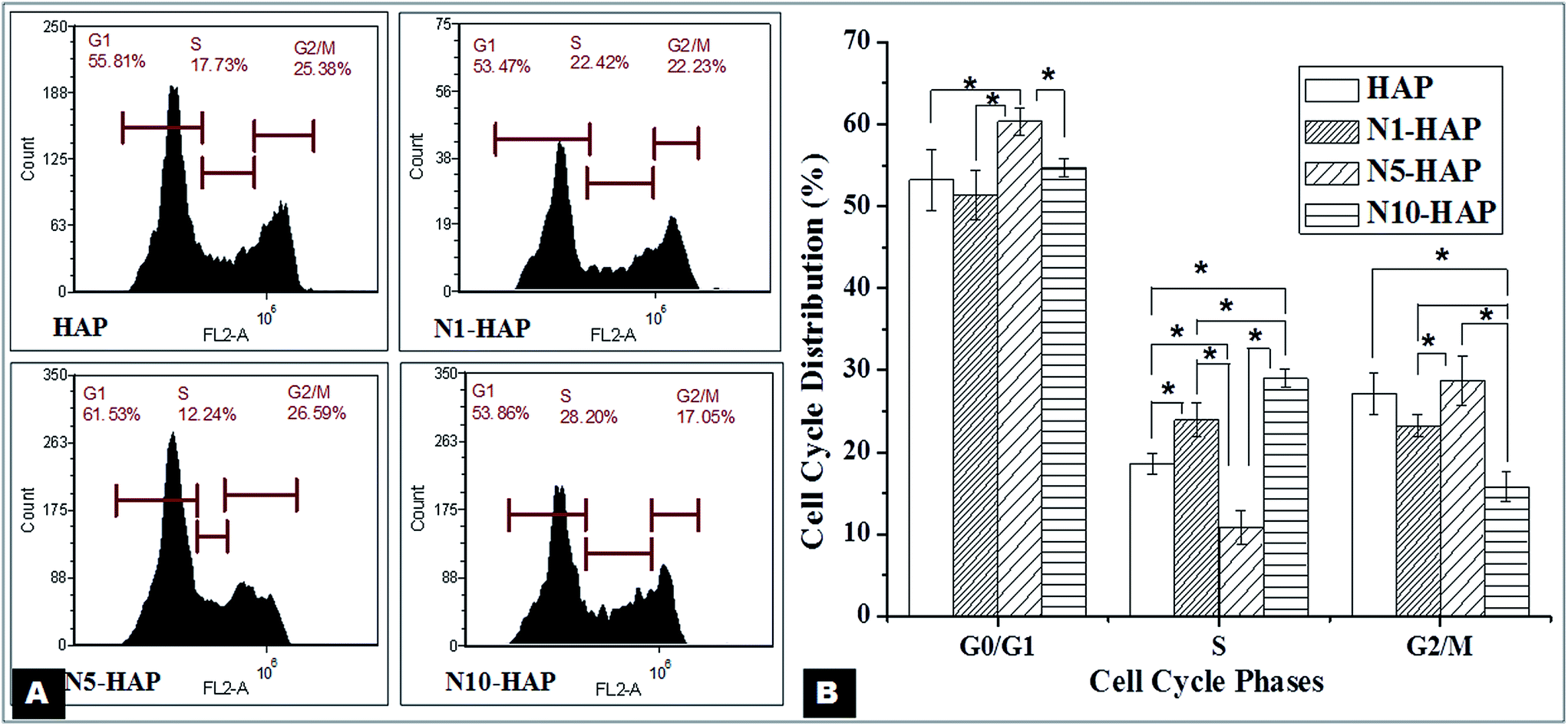 | ||
| Fig. 4 Study of the cell cycle after 24 h of treatment. Cells were treated with Ni2+ doped nHAp at a concentration of 100 μg ml−1 for a period of 24 h. Experiments were done in triplicate. [A] Representative cell cycle histograms for each sample were presented. [B] Quantitative analysis of cell cycle data. Data were expressed as mean ± S.D (n = 3). Statistical significance was checked for p < 0.05. | ||
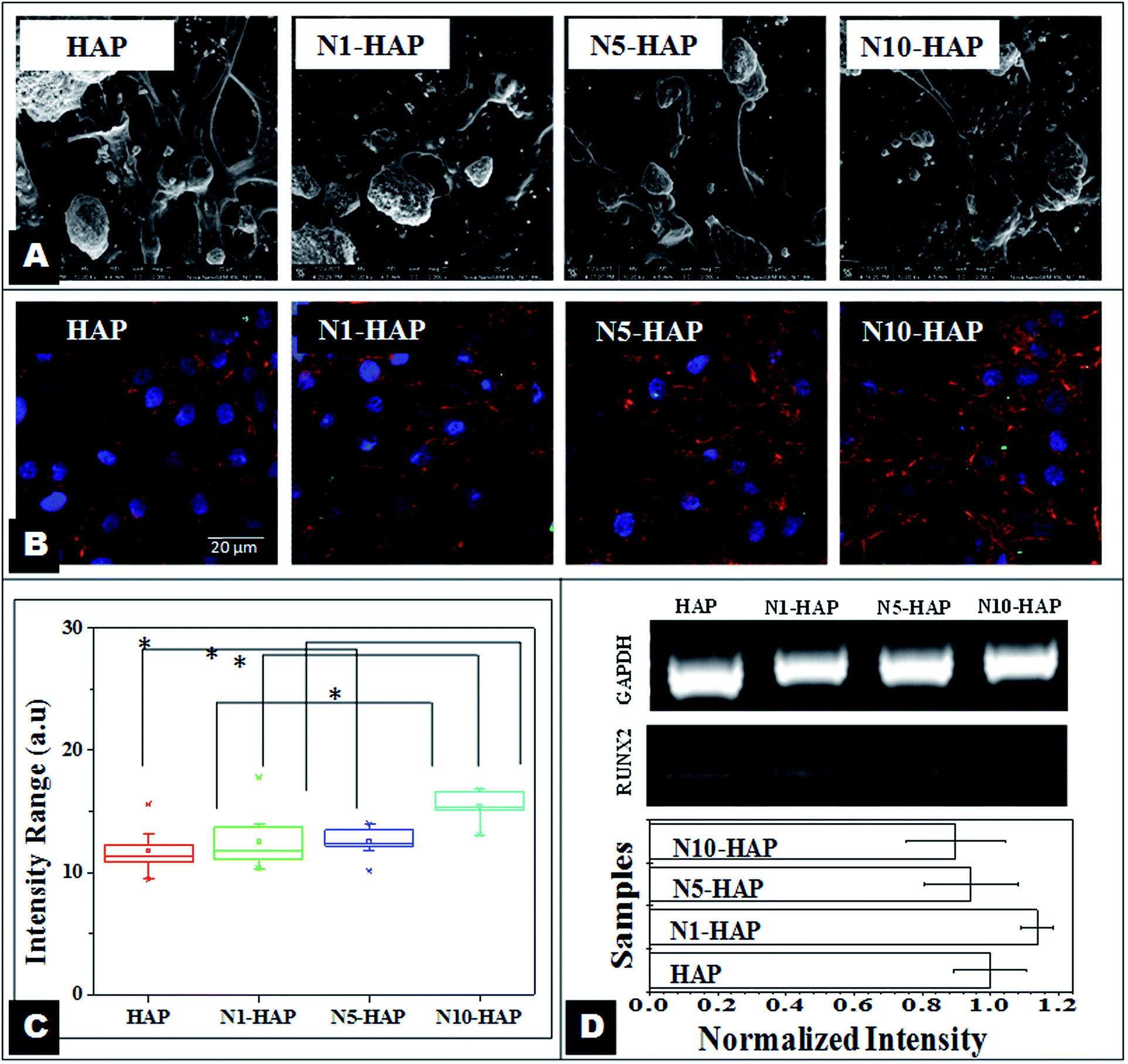 | ||
| Fig. 5 [A] SEM images of cells cultured in presence of nHAp and Ni2+ doped nHAp (at a concentration of 100 μg ml−1) at day 3. [B] Confocal micrographs of cells cultured in presence of nHAp and Ni2+ doped nHAp (at a concentration of 100 μg ml−1) at day 3. Red [F actin], blue [Hoechst] and green [Runx2]. [C] Quantitative image analysis of Runx2 expression. Intensity was calculated for 10 cells. Statistical significance was checked for p < 0.005. [D] Study of Runx2 expression by RT-PCR. Data were expressed as mean ± S.D (n = 3). Statistical significance was checked for p < 0.05. | ||
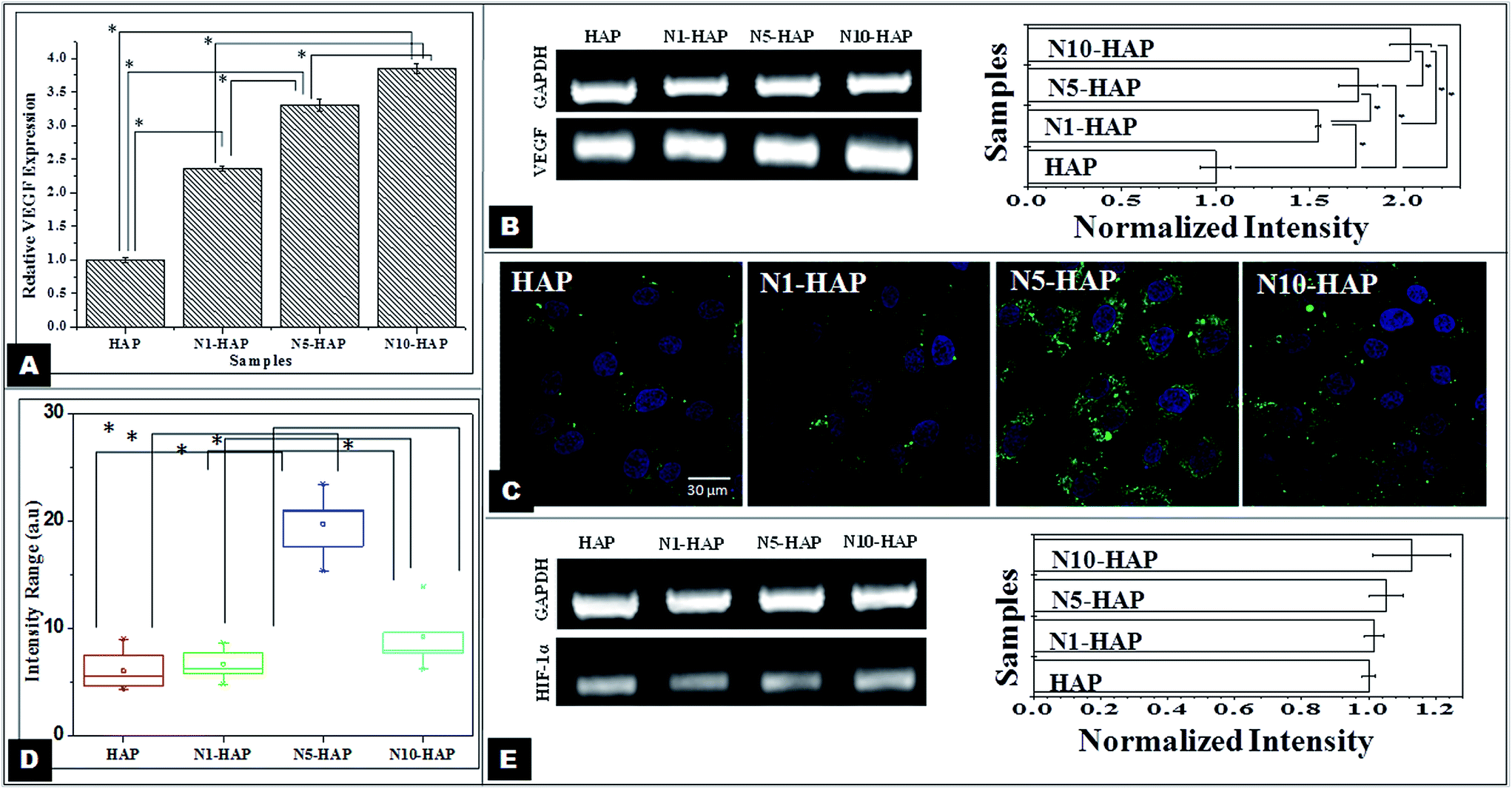 | ||
| Fig. 6 [A] Analysis of VEGF expression by ELISA. Cells were treated with Ni2+ doped nHAp at a concentration of 100 μg ml−1 for a period of 48 h. Data are expressed as mean ± S.D. [B] Study of VEGF mRNA expression by RT-PCR. Data were expressed as mean ± S.D (n = 3). Statistical significance was checked for p < 0.05 [C] Study of HIF-1α expression by MG-63 cells after 12 h post treatment with Ni2+ doped nHAp (at a concentration of 100 μg ml−1) by immuno-cytochemistry. Confocal micrographs of the cells [red (F actin), blue (Hoechst) and green (HIF-1α)]. [D] Quantitative image analysis of HIF-1α expression. Intensity was calculated for 10 cells. Statistical significance was checked for p < 0.005. [E] Study of HIF-1α mRNA expression by RT-PCR. Data were expressed as mean ± S.D (n = 3). Statistical significance was checked for p < 0.05. | ||
4. Discussion
So far only few reports are available on Ni2+ modified HAp. Neelakandeswari et al. (2012) and Wakamura et al. (1998) reported Ni doped HAp synthesis using coprecipitation-ion exchange and microwave assisted technique.26,47 Guerra-López et al. (2000, 2001) previously reported the influence of nickel on calcium HAp crystallization by wet chemical method.48,49 This is the first report on the synthesis of Ni2+ doped nHAp by wet chemical method.Our study showed that yield of Ni2+ doped nHAp was consistent (Table 1) for dopant concentration ranging 1% w/w to 10% w/w. The study showed that the intensity of sample colour increased as the percentage of doping was increased and this may be because of the doping or adsorption of hexaquanickel(II) ion (formed in the reaction solution) to the HAp. A similar observation was reported and explained by Tank et al. (2013) and Yan Li et al. (2010) when they doped Co2+ and Cu2+, respectively in HAp.43,50
The extent of doping is associated with the variation in the functional properties of HAp and metal ion toxicity. ICP-OES results clearly revealed that the actual nickel concentration in the doped HAp was much lower than the toxic concentration. A critical analysis of the doped nHAp (Fig. 1D) revealed that the actual nickel doping was less than that of expected theoretical percentage doping. The variation in actual doping was found significant for all the samples (p < 0.05). In recent report, Guerra-López et al. (2001) demonstrated that the percentage doping in case of transition metals including nickel generally occurs at 1/3rd concentration of the theoretical value.49 This may probably happen due to the formation of stable amine coordination complex of nickel at the basic pH, maintained for the synthesis of nHAp. These complex ions are bigger and bulky, and thus fail to get incorporated into the HAp crystal lattice.47 The size and the shape of the nHAp crystals are very crucial for bone tissue engineering applications. During in vivo bone remodelling, needle like nHAp is synthesized inside the groves of collagen helices. The size of native nHAp crystals varies between (5 nm–20 nm).51 CTAB, being a cationic surfactant, controls the nucleation and particle size of nHAp by providing an in situ miceller soft template.28 In the present study, we also used CTAB during the synthesis for the same purpose. The average size (determined by average ferret diameter) of the synthesized nHAp lied in the range of 14 to 18 nm. The size of N10-HAP particles was found significantly smaller than the other samples (p < 0.05). However, the synthesized nHAp particles were elongated spheroid in shape and not needle-like (Fig. 1A and B). Such deviation in particle shape may occur due to the low concentration of CTAB or high pH of the medium. From FESEM and TEM micrographs (Fig. 1A and B), it was observed that the N10-HAP nanoparticles formed more agglomerates in comparison to other samples, which may be due to its low crystallinity.52 The XRD analysis showed that the sample N10-HAP has percentage crystallinity of only 22% (Table 2), which could be accounted for its higher agglomeration potential. The doping of metal ion in HAp crystal results in the deformation of the crystal structure. Sallam et al. (2012) reported that chromium doping in the HAp led to alteration in the crystal structure as manifested by broadening of the XRD peaks.53 In earlier reports, the Ni2+ doped HAp synthesised by co-precipitation and ion substitution method showed peak broadening.47,54 Interestingly, herein, no such peak broadening was observed, which implies that Ni2+ doping did not distort the apatite crystal much. The minimal variations that occur in the crystal parameter could probably be due the smaller ionic radii of Ni2+ (0.72 Å) in comparison to Ca2+ (0.99 Å). On the contrary, the crystallinity of the nHAp decreased greatly with an increase in the nickel doping (Fig. 2A, Table 2). TEM analysis showed that there was a change in the ‘d’ spacing value corresponding to 002 plane with an increase in the nickel concentration (Table 2) and the difference become approximately 0.01 nm for N10-HAP with respect to HAP.
The FT-IR analysis (Fig. 2B) revealed that with the increase in the doping concentration, the peaks for ν3 stretching (1100–1000 cm−1) and ν2 bending (470 cm−1) of (PO4)−3 became less prominent. These results were found in accordance with Guerra-López et al. (2001) and G. Gergely et al. (2010).49,55 This may be due to the mild variation in the ionic environment of the crystal lattice that leads to an alteration in the stretching field. In addition to the characteristic peaks for the bound water at 1640 and 3450 cm−1, a new peak appeared at 3640 cm−1, attributing to the free OH stretching, which became more intense with the increase in the doping concentration.
The surface area and pore volume play a crucial role in determining the adsorption of biomolecules on the implant. The BET study showed an overall variation of 23% and 35% in surface area and pore volume, respectively, with Ni2+ doping. However, no linear correlation were found between Ni2+ doping and the aforesaid parameters (Table 3). Among the samples, the least surface area and pore volume was found in case of N10-HAP, which may be due to low average particle size of N10-HAP, as evident from TEM analysis.
The zeta potential of the nano materials influences its biological performance in a number of ways including protein adsorption, cell–material interaction and cellular uptake. Doping of the metal ions may change native zeta potential of HAp by altering the surface charge density. At physiological pH, the zeta potential of the synthesized nHAp was found to be negative56 and the metal ion doping results in a variation in its zeta potential. A variation of less than 10 mV was observed with an increase in the nickel concentration from 0 to 10% w/w. Moreover, the increase was not found directly proportional to the nickel concentration. Such low variation in the zeta potential could be accounted by considering low doping (evident from ICP-OES analysis (Fig. 1D)). Our BET analysis also revealed that the highest pore volume for N5-HAP, which may explain the minor anomaly in the zeta potential of N5-HAP. The greater pore size may cause higher entrapment of the positive ions in the pore of the particles, thereby, contributing to the excess zeta.
The protein adsorption onto the implant plays a vital role in determining the host immune response towards the implant.57 Post implantation, a number of plasma proteins including serum albumin, immunoglobins, fibrinogen and fibronectin interact with the biomaterial surface. Hence, BSA was used as a model protein to study the protein adsorbing potential of the Ni2+ doped nHAp. The protein adsorption by nHAp depends on its surface area and zeta potential, more specifically on the charge distribution of Ca/Ni and phosphate ions over the crystal surface, involved in the electrostatic interaction with the proteins.58 Protein adsorption result clearly implied that the doping of nickel did not have any influence on the protein adsorption capacity of the nHAps (Table 3). A critical analysis showed that HAP and N1-HAP had similar surface area and zeta properties, owing to same BSA adsorption. In addition, protein adsorption capacity of N5-HAP was similar to HAP and N1-HAP. However, the zeta potential of N5-HAP was relatively more positive in comparison to others. The pore diameter of nHAp and intermolecular repulsion of BSA may provide a clue for such observation. The average pore diameter for all the samples was less than 1 nm (data not shown) which is much lower than the hydrodynamic radius of BSA. Therefore, BSA molecule cannot be accommodated inside these pores. On the other hand, electrostatic repulsion between two adjacent BSA molecules may also prevent the adsorption of too many BSA molecules on the nHAp surface.
In the context of bone tissue engineering, biomaterials are generally classified either osteoconductive or osteoinductive. Synthetic HAp are found to be osteoconductive, supporting the osteoblast viability and proliferation.59 So far many efforts have been made to improve the osteoconductive property of HAp. Bose et al. (2013) showed an enhanced proliferation of human osteoblast cells in response to treatment with metal ions (Si2+, Sr2+, Mg2+ and Zn2+) doped tri-calcium phosphate TCPs.60 Webster et al. (2010) reported a similar improvement in osteoconductivity of HAp by selective doping of bivalent and trivalent metal ions like Mg2+, Zn2+, La3+, Y3+, In3+, and Bi3+ into the HAp.24 In recent reports, Wu et al. (2012 and 2013) have proven that Cu and Co doped bioglass are highly cytocompatible and support osteoconduction.17,18 Nickel is also a ‘d’ block transition metal and Ryhänen et al. (1997) showed that Ni2+ is osteoconductive in nature.61 As mentioned earlier, Ni has also been used extensively in biomedical implants such as nitinol based stent and dental implants. Our MTT analysis apparently showed that Ni2+ doped nHAp samples help in MG-63 cell proliferation (Fig. 3). Further, to quantify the variation in the live population of cells, flow cytometry based live-dead assay clearly indicated that Ni2+ doped nHAps are cytocompatible (Table 4). Our MTT study showed that there was a decrease in the cell proliferation rate with time. As mentioned earlier, the two possibilities could be either cell death or cell differentiation. It was evident from the live dead assay that there was no significant cell death in the later phase of the culture. On the other hand, cell cycle analysis (Fig. 4) revealed the presence of prolonged G0/G1 phase. In bone physiology, a high population of cells in G0/G1 often indicates their commitment towards differentiation.62 Therefore, a higher value of G0/G1 in the case of N5-HAP treated group could be because of the differentiation potential of the material. FACS analysis also showed that the percentage cell population in pro-G0/G1 (a phase associated with apoptosis) was negligible for all groups.
Previously, Gough et al. (2004) outlined that the formation of the nodule is a signature of osteoblast differentiation.63 FESEM images (Fig. 5A) of bone cells showed that Ni2+ doped materials (N5-HAP and N10-HAP) were supporting nodule formations. There are some signature proteins that can be only seen during the period of differentiation of preosteoblasts to osteocytes like Runx2, osterix and osteocalacin. Transcription factor Runx2 is one of the signature proteins which is a pre-requisite for the osteogenic differentiation of mesenchymal stem cells.64 In this case, a marginal but significant increase in Runx2 expression was observed in case of N10-HAP (Fig. 5B and C). As Runx2 expression can be activated through calcium dependent Wnt5 signalling pathway, bivalent metal ions (Ni2+) can act as agonist to Ca2+ ions and trigger the expression of Runx2.50 It is important to mention that MG-63 is a mature osteoblast. Therefore, Runx2 expression always remains in basal level in MG-63 in comparison to its progenitor. Hence, the average expression of Runx2 was almost similar for all the samples.
ELISA analysis (Fig. 6A and B) showed that the doped samples induced significantly higher VEGF expression in comparison to pure HAP. From the literature, it is evident that among various angiogenic factors such as PDGF, FGF, bEGF, etc.; VEGF plays a key role in signalling pathways pertaining to endothelial cell migration, survival and angiogenesis.65 HIF-1α is one of the key regulators of the VEGF expression.66 The bivalent Ni2+ ions stabilize the HIF-1α, which in turn induce the expression of VEGF. Hence, the Ni2+ doped nHAp samples showed better expression of VEGF. In the similar studies, Wu et al. (2012 & 2013) showed that bivalent metal ions (cobalt and copper) incorporated mesoporous bioglasses induced higher level of VEGF expression in human bone marrow derived stromal cells. However, comparison of these results showed 3.8 fold increase in VEGF production for 3.22% nickel doping (the actual Ni doping in N10-HAP) in comparison to undoped ceramics, while the same was 1.2 fold for 5% cobalt doping in cobalt doped bioglass.17,18 This clearly suggest that our Ni2+ doped nHAp can induce VEGF more effectively with lower concentration, hence reduce the health risk.
In the present study, the immunocytochemistry profile of HIF-1α showed that the nickel doped HAp samples had a significantly higher expression of HIF-1α (Fig. 6C and D). Wang et al. (2007) and Malda et al. (2007) showed that HIF-1α, a key regulator of the angiogenic genes, expressed during the bone repair. During morphogenesis, vascularization of the bone callus is the key step for bone formation.10,67 Bivalent metal ions such as nickel, cobalt, and copper, etc. mimics hypoxia condition by replacing Fe2+ ions, the key cofactor of the PH enzyme and thereby, inactivating the enzyme and stabilizing HIF-1α. Interestingly, in this study, we observed that expression of HIF-1α did not show any linear correlation with the nickel doping concentration. In case of HIF-1α expression, N5-HAP showed the highest expression followed by N10-HAP, N1-HAP and HAP, respectively. However, in case of VEGF, the increase in the expression was linear with respect to nickel doping concentration. It was also reported for copper and cobalt doped proangiogenic biomaterials, the VEGF expression profile does not always follow the HIF-1α profile. This might be explained on the basis that VEGF expression does not merely depend on the HIF-1α expression level, but also on the mitogen-activated protein (MAP) kinase activity and subsequent phosphorylation mediated activation of HIF-1α. Hence N10-HAP, which might have a sub-optimal HIF-1α expression followed by better MAP kinases activity68 and phosphorylated activation of HIF-1α, resulted in better VEGF expression. Another reason could be the interplay of Runx2 with HIF-1α which is supposedly stabilizing HIF-1α by competing with the Von HippelLindau Protein (pVHL).69 From the Runx2 immunocytochemistry expression profile, N5-HAP showed better expression of Runx2 followed by N10-HAP. Such higher expression of Runx2 in case of N5-HAP may contribute to higher expression of HIF-1α. In a similar study earlier, we showed that angiogenic properties of HAp can be improved by dual doping of Co2+ and Mg2+.70
All the results pertaining to the characterization of the effect of Ni2+ doped nHAp on MG-63 cells indicate that Ni2+ doped nHAp can be used as a VEGF inducing osteoconductive biomaterials for bone tissue engineering. However it is important to mention that although MG-63 is often used as osteoblastic model, but it is actually an osteosarcoma cell line derived from malignant bone tumor. Earlier studies have shown that response of MG-63 towards a biomaterials (interms of proliferation, migration and composition of synthesized ECM) often differed in comparison to that of primary human osteoblast.71,72 Hence, performance of Ni2+ doped nHAp should also be evaluated using primary human osteoblast cells before preclinical study.
5. Conclusion
Our study showed that Ni2+ doped nHAp particles of narrow size distribution can effectively be prepared by wet chemical precipitation method using CTAB. Doping of Ni2+ upto 2.97% w/w of Ca2+ did not cause crystal deformation. Similarly, physical properties like surface area, zeta potential and protein adsorption remained almost same with Ni2+ doping. All the doped samples were bone cell compatible and osteoconductive. Analysis showed that Ni2+ doped nHAp samples were potent inducer of cellular VEGF and the expression of VEGF was directly proportional to the doping concentration of Ni2+. A mechanistic analysis further implied involvement of Ni2+ in HIF-1α stabilization followed by increased VEGF expression. Comparison of these results with the relevant literature revealed that the relative VEGF expression per unit quantity of doping was higher in the present case.In conclusion, Ni2+ doped nHAp is a proangiogenic–osteogenic material and could be used in bone tissue engineering. Response of endothelial cells (viability and tube formation) and primary osteoblast against these materials in vitro has to be studied. Moreover, it is important to assess the biological performance of the materials in vivo.
Acknowledgements
1. PSG Institute of Advanced Studies for HR-TEM analysis. 2. SAIF, IIT Madras for ICP-OES analysis. 3. DBT, Govt of India (RGYI scheme project no. BT/PR6230/GBD/27/391/2012). 4. DBT, Govt of India (RGYI scheme project no. BT/PR6227/GBD/27/390/2012). 5. Prof. T. K. Maiti for providing the molecular biology laboratory facility. 6. Centre of Excellence, orthopaedic Tissue engineering and Rehabilitation (TEQIP–II).References
- M. Frohlich, W. L. Grayson, L. Q. Wan, D. Marolt, M. Drobnic and G. Vunjak-Novakovic, Curr. Stem Cell Res. Ther., 2008, 3, 254–264 CrossRef CAS.
- M. Mravic, B. Péault and A. W. James, BioMed Res. Int., 2014, 2014, 1–5 CrossRef PubMed.
- A. Perets, Y. Baruch, F. Weisbuch, G. Shoshany, G. Neufeld and S. Cohen, J. Biomed. Mater. Res., Part A, 2003, 65, 489–497 CrossRef PubMed.
- M. Sheridan, L. Shea, M. Peters and D. Mooney, J. Controlled Release, 2000, 64, 91–102 CrossRef CAS.
- W. D. Holder Jr, H. E. Gruber, W. D. Roland, A. L. Moore, C. R. Culberson, A. B. Loebsack, K. J. Burg and D. J. Mooney, Tissue Eng., 1997, 3, 149–160 CrossRef.
- A. Hasan, A. Paul, N. E. Vrana, X. Zhao, A. Memic, Y.-S. Hwang, M. R. Dokmeci and A. Khademhosseini, Biomaterials, 2014, 35, 7308–7325 CrossRef CAS PubMed.
- S. Baiguera and D. Ribatti, Angiogenesis, 2013, 16, 1–14 CrossRef CAS PubMed.
- R. C. Fields, A. Solan, K. T. McDonagh, L. E. Niklason and J. H. Lawson, Tissue Eng., 2003, 9, 1281–1287 CrossRef CAS PubMed.
- H. Iwaguro, J.-I. Yamaguchi, C. Kalka, S. Murasawa, H. Masuda, S.-I. Hayashi, M. Silver, T. Li, J. M. Isner and T. Asahara, Circulation, 2002, 105, 732–738 CrossRef CAS PubMed.
- Y. Wang, C. Wan, L. Deng, X. Liu, X. Cao, S. R. Gilbert, M. L. Bouxsein, M.-C. Faugere, R. E. Guldberg and L. C. Gerstenfeld, J. Clin. Invest., 2007, 117, 1616 CAS.
- Y.-S. Chun, M.-S. Kim and J.-W. Park, J. Korean Med. Sci., 2002, 17, 581 CrossRef CAS.
- G. Chachami, G. Simos, A. Hatziefthimiou, S. Bonanou, P.-A. Molyvdas and E. Paraskeva, Am. J. Respir. Cell Mol. Biol., 2004, 31, 544–551 CrossRef CAS PubMed.
- W. Feng, F. Ye, W. Xue, Z. Zhou and Y. J. Kang, Mol. Pharmacol., 2009, 75, 174–182 CrossRef CAS.
- K. Vandamme, X. Holy, M. Bensidhoum, D. Logeart-Avramoglou, I. Naert, J. Duyck and H. Petite, Tissue Eng., Part C, 2010, 17, 311–318 CrossRef PubMed.
- J.-T. Hwang, M. Lee, S.-N. Jung, H.-J. Lee, I. Kang, S.-S. Kim and J. Ha, Carcinogenesis, 2004, 25, 2497–2507 CrossRef CAS PubMed.
- M. Kaczmarek, O. A. Timofeeva, A. Karaczyn, A. Malyguine, K. S. Kasprzak and K. Salnikow, Free Radicals Biol. Med., 2007, 42, 1246–1257 CrossRef CAS PubMed.
- C. Wu, Y. Zhou, W. Fan, P. Han, J. Chang, J. Yuen, M. Zhang and Y. Xiao, Biomaterials, 2012, 33, 2076–2085 CrossRef CAS PubMed.
- C. Wu, Y. Zhou, M. Xu, P. Han, L. Chen, J. Chang and Y. Xiao, Biomaterials, 2013, 34, 422–433 CrossRef CAS PubMed.
- K. Das, S. Das and S. Dhundasi, Indian J. Med. Res., 2008, 128, 412 CAS.
- K. Salnikow, W. G. An, G. Melillo, M. V. Blagosklonny and M. Costa, Carcinogenesis, 1999, 20, 1819–1823 CrossRef CAS PubMed.
- S. Civjan, E. F. Huget and L. B. DeSimon, J. Dent. Res., 1975, 54, 89–96 CrossRef CAS PubMed.
- M. Fini, N. N. Aldini, P. Torricelli, G. Giavaresi, V. Borsari, H. Lenger, J. Bernauer, R. Giardino, R. Chiesa and A. Cigada, Biomaterials, 2003, 24, 4929–4939 CrossRef CAS.
- A. A. Nemec and A. Barchowsky, FASEB J., 2008, 22, 764 Search PubMed.
- T. J. Webster, E. A. Massa-Schlueter, J. L. Smith and E. B. Slamovich, Biomaterials, 2004, 25, 2111–2121 CrossRef CAS PubMed.
- H. Lukman, Z. Yaakob, I. Manal and W. R. Wan Daud, Key Eng. Mater., 2010, 447, 770–774 CrossRef.
- N. Neelakandeswari, G. Sangami, P. Emayavaramban, R. Karvembu, N. Dharmaraj and H. Y. Kim, Tetrahedron Lett., 2012, 53, 2980–2984 CrossRef CAS PubMed.
- N. Monmaturapoj, J. Met., Mater. Miner., 2008, 18, 15–20 CAS.
- M. Salarian, M. Solati-Hashjin, S. S. Shafiei, A. Goudarzi, R. Salarian and A. Nemati, Mater. Sci., 2009, 27, 961–971 CAS.
- T. Liu, Y. Yan and S. Li, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2008, 23, 395–398 CrossRef.
- K. Sanosh, M.-C. Chu, A. Balakrishnan, T. Kim and S.-J. Cho, Bull. Mater. Sci., 2009, 32, 465–470 CrossRef CAS.
- S. Tay, A. Haseeb, M. R. Johan, P. Munroe and M. Quadir, Intermetallics, 2013, 33, 8–15 CrossRef CAS PubMed.
- M. Wadekar, C. Rode, Y. Bendale, K. Patil, A. Gaikwad and A. Prabhune, J. Pharm. Biomed. Anal., 2006, 41, 1473–1478 CrossRef CAS PubMed.
- M. Saeri, A. Afshar, M. Ghorbani, N. Ehsani and C. Sorrell, Mater. Lett., 2003, 57, 4064–4069 CrossRef CAS.
- N. Puvvada, P. K. Panigrahi and A. Pathak, Nanoscale, 2010, 2, 2631–2638 RSC.
- L. Chen, J. M. Mccrate, J. C. Lee and H. Li, Nanotechnology, 2011, 22, 105708 CrossRef PubMed.
- D. T. H. Wassell, R. C. Hall and G. Embery, Biomaterials, 1995, 16, 697–702 CrossRef CAS.
- S. Panseri, C. Cunha, T. D'Alessandro, M. Sandri, G. Giavaresi, M. Marcacci, C. T. Hung and A. Tampieri, J. Nanobiotechnol., 2012, 10, 32 CrossRef CAS PubMed.
- K. Cheng, W. Weng, H. Wang and S. Zhang, Biomaterials, 2005, 26, 6288–6295 CrossRef CAS PubMed.
- Z. Shi, X. Huang, Y. Cai, R. Tang and D. Yang, Acta Biomater., 2009, 5, 338–345 CrossRef CAS PubMed.
- I. R. Zerbo, A. L. Bronckers, G. de Lange and E. H. Burger, Biomaterials, 2005, 26, 1445–1451 CrossRef CAS.
- D. Armstrong, A. J. Augustin, R. Spengler, A. Al-Jada, T. Nickola, F. Grus and F. Koch, Ophthalmologica, 1997, 212, 410–414 CrossRef PubMed.
- M. Sadat-Shojai, M.-T. Khorasani, E. Dinpanah-Khoshdargi and A. Jamshidi, Acta Biomater., 2013, 9, 7591–7621 CrossRef CAS PubMed.
- K. P. Tank, K. S. Chudasama, V. S. Thaker and M. J. Joshi, J. Nanopart. Res., 2013, 15, 1–11 CrossRef.
- Y. Han, X. Wang and S. Li, J. Nanopart. Res., 2009, 11, 1235–1240 CrossRef CAS.
- Z. Yan-Zhong, H. Yan-Yan, Z. Jun, Z. Shai-Hong, L. Zhi-You and Z. Ke-Chao, Nanoscale Res. Lett., 2011, 6, 1–8 CrossRef PubMed.
- A. Rajendran, R. C. Barik, D. Natarajan, M. Kiran and D. K. Pattanayak, Ceram. Int., 2014, 40, 10831–10838 CrossRef CAS PubMed.
- M. Wakamura, K. Kandori and T. Ishikawa, Colloids Surf., A, 1998, 142, 107–116 CrossRef CAS.
- J. Guerra-López, R. González, A. Gómez, R. Pomés, G. Punte and C. Della Védova, J. Solid State Chem., 2000, 151, 163–169 CrossRef.
- J. Guerra-López, R. Pomes, C. Della Védova, R. Vina and G. Punte, J. Raman Spectrosc., 2001, 32, 255–261 CrossRef PubMed.
- Y. Li, J. Ho and C. P. Ooi, Mater. Sci. Eng., C, 2010, 30, 1137–1144 CrossRef CAS PubMed.
- M. Ferraz, F. Monteiro and C. Manuel, J. Appl. Biomater. Biomech., 2004, 2, 74–80 CAS.
- Y.-J. Shyong, R.-F. Lin, H.-S. Soung, H.-H. Wei, Y.-S. Hsueh, K.-C. Chang and F.-H. Lin, J. Mater. Chem. B, 2015, 3, 2331–2340 RSC.
- S. M. Sallam, K. M. Tohami, A. M. Sallam, L. I. A. Salem and F. A. Mohamed, J. Biophys. Chem., 2012, 3, 278–282 CrossRef.
- G. Mabilleau, R. Filmon, P. Petrov, M. Baslé, A. Sabokbar and D. Chappard, Acta Biomater., 2010, 6, 1555–1560 CrossRef CAS PubMed.
- G. Gergely, F. Wéber, I. Lukács, L. Illés, A. L. Tóth, Z. E. Horváth, J. Mihály and C. Balázsi, Cent. Eur. J. Chem., 2010, 8, 375–381 CrossRef CAS.
- R. L. Williams, M. J. Hadley, P. J. Jiang, N. A. Rowson, P. M. Mendes, J. Z. Rappoport and L. M. Grover, J. Mater. Chem. B, 2013, 1, 4370–4378 RSC.
- J. M. Anderson, A. Rodriguez and D. T. Chang, Semin. Immunol., 2008, 20, 86–100 CrossRef CAS PubMed.
- Z. Zhuang, T. J. Fujimi, M. Nakamura, T. Konishi, H. Yoshimura and M. Aizawa, Acta Biomater., 2013, 9, 6732–6740 CrossRef CAS.
- R. Z. LeGeros, Clin. Orthop. Relat. Res., 2002, 395, 81–98 CrossRef.
- S. Bose, G. Fielding, S. Tarafder and A. Bandyopadhyay, Trends Biotechnol., 2013, 31, 594–605 CrossRef CAS PubMed.
- J. Ryhänen, E. Niemi, W. Serlo, E. Niemelä, P. Sandvik, H. Pernu and T. Salo, J. Biomed. Mater. Res., 1997, 35, 451–457 CrossRef.
- M. Carroll, Y. Zhu and A. D. D'Andrea, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 2869–2873 CrossRef CAS.
- J. E. Gough, J. R. Jones and L. L. Hench, Biomaterials, 2004, 25, 2039–2046 CrossRef CAS PubMed.
- T. Komori, Front. Biosci., 2007, 13, 898–903 CrossRef PubMed.
- M. Shibuya, Cell Struct. Funct., 2001, 26, 25–35 CrossRef CAS.
- D. Shweiki, A. Itin, D. Soffer and E. Keshet, Nature, 1992, 359, 843–845 CrossRef CAS PubMed.
- J. Malda, T. J. Klein and Z. Upton, Tissue Eng., 2007, 13, 2153–2162 CrossRef CAS PubMed.
- Q. Ke and M. Costa, Mol. Pharmacol., 2006, 70, 1469–1480 CrossRef CAS PubMed.
- S.-H. Lee, X. Che, J.-H. Jeong, J.-Y. Choi, Y.-J. Lee, Y.-H. Lee, S.-C. Bae and Y.-M. Lee, J. Biol. Chem., 2012, 287, 14760–14771 CrossRef CAS PubMed.
- S. Kulanthaivel, U. Mishra, T. Agarwal, S. Giri, K. Pal, K. Pramanik and I. Banerjee, Ceram. Int., 2015, 41, 11323–11333 CrossRef CAS PubMed.
- C. Pautke, M. Schieker, T. Tischer, A. Kolk, P. Neth, W. Mutschler and S. Milz, Anticancer Res., 2004, 24, 3743–3748 CAS.
- A. Perez, R. Spears, J. Gutmann and L. Opperman, Int. Endod. J., 2003, 36, 564–570 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra09560c |
| This journal is © The Royal Society of Chemistry 2015 |