
Novel flame retardant flexible polyurethane foam: plasma induced graft-polymerization of phosphonates
Abstract
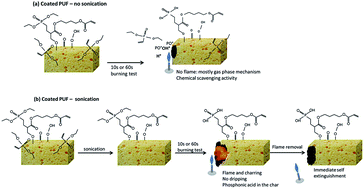
Flame retardancy of flexible polyurethane foams has become an issue due to very severe regulations. To overcome the challenge of incorporating flame retardant (FR) additives during the foaming process without altering the foam properties, a plasma surface treatment was used for the first time in this work: a cold plasma induced graft-polymerization of phosphonate containing precursors (diethylvinylphosphonate-DEVP) with or without a crosslinking agent (1,4 butanedioldiacrylate) was applied on open cell flexible polyurethane foams (PUF). The flame retardant properties of these foams, before and after further rinsing, were evaluated using horizontal UL-94 test for 10 s and 60 s. One of the tested systems (DEVP + crosslinker), even after sonication, completely stops the melt dripping of the foams when exposed to the flame of the butane torch and charring occurs. This efficient surface treatment was characterized before and after burning by Scanning Electron Microscopy (SEM), electron probe microanalysis (EPMA), 31P solid state NMR and X-ray Photoelectron Spectroscopy (XPS). Its FR mechanism of action was then further investigated using microscale combustion calorimetry (MCC), thermogravimetric analyses coupled with infrared Fourier transform spectroscopy (TGA-FTIR), pyrolysis gas-chromatography coupled with mass spectrometry (GC-MS pyrolysis) and 31P solid state NMR. The results obtained show that it is necessary (i) to use the crosslinking agent, as DEVP mainly reacts with this crosslinker and (ii) to activate the PU foam before graft-polymerization to promote further reaction with the crosslinker. The characterizations also proved that before sonication, non-reacted DEVP precursors mainly act in the gas phase, preventing ignition, whereas after sonication the covalently grafted phosphorus containing species mainly act in the condensed phase to form phosphonic acid which will promote charring and thus will limit dripping and flame spread.
 Please wait while we load your content...
Please wait while we load your content...

