
A straightforward and “green” solvothermal synthesis of Al doped zinc oxide plasmonic nanocrystals and piezoresistive elastomer nanocomposite
Abstract
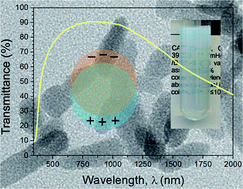
Plasmonic oxide nanocrystals hold great promise in a wide range of applications, for which the availability of scalable and “green” synthesis methods is prerequisite, whereas until recently an excellent response has been demonstrated only for samples prepared through intricate synthesis paths. We report here a simple ethanol solvothermal synthesis route of Al doped ZnO plasmonic nanocrystals (Zn1−xAlxO) at doping levels of x up to 0.15. The obtained Al doped ZnO samples consisted of nanoparticles and short nanorods with a diameter of around 10 nm at x = 0.15 doping level while reaching aspect ratio levels of 50 for lower doping levels. Detailed structural studies using powder X-ray diffraction Rietveld refinement, X-ray absorption and photoelectron spectroscopies show that all samples maintain the structure of the phase-pure zincite with the space group P63mc. The resulting powders exhibit strong infrared absorption while remaining largely transparent for visible light, enabling the preparation of transparent colloidal dispersions. Furthermore, as a test of applicability in a practical device, the nanocrystals were used to prepare transparent piezoresistive Zn0.925Al0.075O–polydimethylsiloxane composites. The prepared sensor material exhibits excellent repeatable and reproducible piezoresistive behaviour.
 Please wait while we load your content...
Please wait while we load your content...

