
Kinetic and thermodynamic study of polyaniline functionalized magnetic mesoporous silica for magnetic field guided dye adsorption†
Abstract
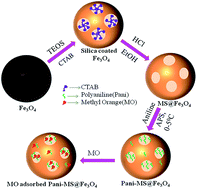
An eco-friendly magnetic mesoporous silica iron oxide (MS@Fe3O4) nanoparticles with a high surface area was fabricated using a colloidal chemical method. Hereafter, polyaniline (PANI) was conjugated into the pores of MS@Fe3O4 to obtain PANI–MS@Fe3O4 nanocomposites. The nanocomposites were essentially monodispersed and highly efficient for the adsorption of methyl orange (MO). In addition, the rate of the adsorption reaction followed pseudo-second-order kinetics and the adsorption isotherm fitted well to the Langmuir isotherm model. The negative values for the change in Gibbs energy (ΔG) and positive value of the change in enthalpy (ΔH) indicated that the overall adsorption process was spontaneous and endothermic in nature. The density functional theory calculation using the Gaussian 09 and GaussView 5.0 programs also supported the electrostatic interaction phenomena between PANI and MO, which is mainly responsible for the adsorption.
 Please wait while we load your content...
Please wait while we load your content...

