
Synthesis, characterization and micellization of amphiphilic polyethylene-b-polyphosphoester block copolymers†
Abstract
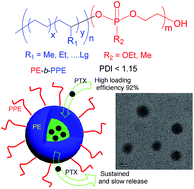
A sequential synthetic strategy combining metal catalyzed living polymerization of ethylene and ring-opening polymerization (ROP) was successfully used to prepare novel polyethylene-block-polyphosphoester (PE-b-PPE) diblock copolymers. Narrowly dispersed hydroxyl-terminated polyethylene (PE-OH) was first synthesized by amine–imine nickel catalyzed-living polymerization of ethylene and a subsequent chain transfer reaction. PE-b-PPE diblock copolymers with narrow polydispersities were obtained by organocatalyzed ring-opening polymerization (ROP) of 2-ethyoxyl-2-oxo-1,3,2-dioxaphospholane or 2-methyl-2-oxo-1,3,2-dioxaphospholane using PE-OH as a macroinitiator. Investigations of self-assembly of the obtained PE-b-PPEs block copolymers in water by means of TEM and laser light scattering confirmed that the amphiphilic block copolymers formed micelles in aqueous solution. These PE-b-PPE block copolymers were used for paclitaxel (PTX) encapsulation to demonstrate their potential in drug delivery. These biocompatible PE-PPE polymeric micelles showed a high loading efficiency of 92% and content of PTX of 16.6 wt% as well as slow and sustained release behavior.
 Please wait while we load your content...
Please wait while we load your content...

