
Mixed amido-cyclopentadienyl group 4 metal complexes†
Aleš
Havlík
ab,
Martin
Lamač
a,
Jiří
Pinkas
a,
Aleš
Růžička
b and
Michal
Horáček
*a
aJ. Heyrovský Institute of Physical Chemistry, Academy of Sciences of The Czech Republic, v.v.i., Dolejškova 3, 182 23 Prague 8, Czech Republic. E-mail: michal.horacek@jh-inst.cas.cz; Fax: +420 28658 2307; Tel: +420 26605 3965
bDepartment of General and Inorganic Chemistry, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 532 10 Pardubice 2, Czech Republic
First published on 2nd July 2015
Abstract
The reactivity of substituted half-sandwich complexes of Ti, Zr, and Hf towards 2,6-diisopropylaniline NH2C6H3-2,6-iPr2 (LNH) and 2-[(dimethylamino)methyl]aniline NH2C6H4-2-(CH2NMe2) (LNNH) was investigated. Two series of mononuclear complexes (η5-C5Me5)LNMCl2 (M = Ti (3), Zr (4), Hf (5)) and (η5-C5Me5)LNNMCl2 (M = Ti (7), Zr (8), Hf (9)) were prepared from corresponding (pentamethylcyclopentadienyl)metal trichlorides and the lithium precursors LNLi and LNNLi. Besides the desired products a dimeric hafnium compound [(η5-C5Me5){µ-NC6H4-2-(CH2NMe2)}HfCl]2 (10) was also isolated and structurally characterized. The formation of dimeric complexes of this type was also achieved by reacting a series of (η5-C5Me4R)LCNMCl2 (M = Zr, Hf; R = Me, H; LCN = C6H4-2-(CH2NMe2)-µ2C,N) complexes with lithium precursors LNLi and LNNLi generating [(η5-C5Me4H){µ-NC6H3-2,6-iPr2}MCl]2 (M = Zr (11), Hf (13)), [(η5-C5Me5){µ-NC6H3-2,6-iPr2}MCl]2 (M = Zr (12), Hf (14)), [(η5-C5Me4H){µ-NC6H4-2-(CH2NMe2)-µ2N,N}ZrCl]2 (15), and [(η5-C5Me5){µ-NC6H4-2-(CH2NMe2)}ZrCl]2 (16), respectively. The formation of 10 by this reaction procedure was not detected; instead monomeric complexes (η5-C5Me4R)LNNLCNHfCl (R = H (17), Me (18)) were observed as major products. NMR and IR spectroscopy techniques and elemental analysis were used for characterization of the prepared complexes, whereas the structures in most cases were determined by X-ray crystallography.
Introduction
The significant interest of several research groups in the area of half-sandwich cyclopentadienyl–amino, –amido, and –imido substituted group 4 compounds is driven mainly by their applications in catalytic transformations of unsaturated compounds.1 The development of new imido-supported non-metallocene olefin polymerization catalysts seems to be a popular area at the present time.2Earlier works in the field of early transition metal amide and imide complexes were reviewed by Nugent and Haymore3 and Gade and Mountford.4 In the latter article, structures of titanium complexes bearing ligands with adjacent donor functional groups and their reactivity towards unsaturated compounds are described. Mindiola5 summarized the work on novel metal–ligand multiply bonded archetypes including the M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NR6 one which could be described as a nitrene group (:NR) and which parallels the metal–carbon double bond functionality (carbene:CR2). The preparation of these complexes is connected to the oxidatively induced α-hydrogen deprotonation of metal-bound amide in low valent titanium species. Such compounds are acting in various catalytic processes such as hydroamination, hydrohydrazination, aziridination, multicomponent coupling reactions, guanylations, and carboamination. For example, titanium hydrazido and imido complexes containing a pyrrolyl-based ligand are active in hydroamination of alkynes.7
NR6 one which could be described as a nitrene group (:NR) and which parallels the metal–carbon double bond functionality (carbene:CR2). The preparation of these complexes is connected to the oxidatively induced α-hydrogen deprotonation of metal-bound amide in low valent titanium species. Such compounds are acting in various catalytic processes such as hydroamination, hydrohydrazination, aziridination, multicomponent coupling reactions, guanylations, and carboamination. For example, titanium hydrazido and imido complexes containing a pyrrolyl-based ligand are active in hydroamination of alkynes.7
Mono- and dinuclear hafnium imido complexes containing bulky Cp* (Cp* = η5-pentamethylcyclopentadienyl) and arylsilyl ligands were prepared by Tilley.8 Continuation of work on this topic (for titanium) with simpler and improved synthetic procedures and a plethora of structure descriptions of titanium imides were reported by Lorber.9 The homo- and heterobimetallic complexes of Ti, Zr, Hf, V, and Mo with amido and imido ligands in both bridging and terminal positions were isolated recently.10 In the Ti/Mo heterobimetallic systems the η6-arene-coordinated Mo atom can act as a central atom in a Fischer-type carbene.10b Structures of amide–chloride and amide–imide complexes of group 4 metals containing rather bulky hexamethyldisilazide ligands with cyclic as well as acyclic frameworks were also determined.11
Ligands bearing additional functional groups attached to the amide/imide moiety were also applied. For example, phosphine-substituted anilines were used by Bochmann for the preparation of imido bridged titanium complexes incorporating the central four membered Ti2N2 ring. The presence of adjacent phosphine donor groups was further utilized for the complexation of platinum metals. Moreover, the titanium complexes revealed promising activity in the polymerization of ethylene without a change of the central Ti2N2 framework upon activation by MAO (methylaluminoxane).12 Both amido- and imidoethylpyridine and aniline N-bridged titanium complexes with the pendant and potentially chelating N-groups ([N(R)(CH2CH2py)], py = C5H4N, R = SiMe3, SitBuMe2, Ph) were described by Martins.13
The chemistry of group 4 amides or imides containing a η5-cyclopentadienyl or related ring is rather rich in structural motifs. These motifs are represented by the amide Cp*TiCl2NH-tBu,14 bis-amides [N(Me)C6H3-2,6-iPr2]2Cp*ZrCl,15 [(η5-1,3-Me2C5H2)Ti(NMe2)2Cl]–CH2–[(η5-C9H9)Ti(NMe2)2Cl],16 and CpTiCl(NMe2)2 (ref. 17) (Cp = η5-cyclopentadienyl), tris-amides Cp*Zr(NH-tBu)3 and Cp*Hf(LN)3,18 and chelating bis-amides.19 Dinuclear mono- and dicyclopentadienyl complexes with four-membered symmetrical rings formed of two metal atoms bridged usually by two imido nitrogen atoms are the dominant group of complexes in this field.8,18,20 Other compounds of similar constitution are the products of nitrogen activation by a low-valent metallocene moiety.21 Higher nuclearity or cyclicity compounds are also known.21i,21j,22
Being interested in the preparation and potential use of bifunctional compounds of mainly group 4 metals, we described in our previous work a simple method for the preparation of compounds containing a highly substituted cyclopentadienyl ring and the C,N-chelating ligand C6H4-2-(CH2NMe2)-µ2C,N (LCN)23 or bifunctional β-diketiminate ligands with the potentially coordinating methoxy groups.24 When we tried to introduce another sterically demanding substituent like 2,6-diisopropylaniline into the coordination sphere of these metal ions the formation of dimeric compounds with arylimide bridges accompanied by release of C,N-chelating ligand were observed. These unexpected results led us to perform a comparative study of such a phenomenon using a sterically demanding as well as N,N-chelating anilines.
Results and discussion
The complexes (η5-C5Me5)MCl3 (M = Ti, Zr, Hf) react smoothly with one equivalent of LNLi (1) to afford complexes (η5-C5Me5)LNMCl2 (M = Ti (3), Zr (4), Hf (5)) and LiCl (Scheme 1). | ||
| Scheme 1 Preparation of compounds 3–5. | ||
A difference in long-term stability of the prepared amide complexes was observed. Whereas the isolable yield of Zr and Hf complexes was more than 50%, the Ti complex could not be isolated sufficiently pure in larger amount due to its decomposition. NMR spectra of the crude product were, however, in line with the proposed structure, notably displaying a signal due to the N–H hydrogen at δ 9.51 ppm. Moreover, we were able to isolate several crystals of 3 suitable for X-ray analysis directly from the cooled reaction mixture. The molecular structure of 3 obtained by X-ray analysis showed that the expected product was formed. Nevertheless, further processing of the crude product led to the formation of a mixture of yet unidentified compounds which do not contain N–H bond (according to NMR and IR spectroscopy). This could be due to the thermolytic α-hydrogen abstraction from the metal-bound amide by a chlorine atom as described by Tilley for hafnium complexes.8
In contrast, zirconium and hafnium complexes 4 and 5 are stable and were isolated and characterized by NMR and IR spectroscopy. 1H and 13C NMR spectra of amide complexes 4 and 5 supported the proposed structures. The signal of the amine hydrogen is downfield shifted (6.37 ppm for 4 and 6.02 for 5) in comparison to the free 2,6-diisopropylaniline ligand (δH NH (TOL-d8): 3.18 ppm). Infrared spectra of 4 and 5 complexes displayed absorption bands corresponding to N–H stretching vibration at 3352 cm−1 for 4 and 3355 cm−1 for 5. There are also present the aromatic C–H stretching vibration at 3062 cm−1 and strong absorption for C–H out of plane deformation bands at 750 cm−1 typical for an ortho-substituted benzene ligand. The asymmetric stretching vibration of methyl groups in Cp* ligands were observed at 2962–2869 cm−1, and their asymmetric deformation vibration in range 1460–1430 cm−1. The strong deformation vibration corresponding to methyl groups in 2,6-diisopropylaniline ligand were found at 1380 cm−1.
In addition, minor product (η5-C5Me5)(LNN)3Zr (6) was obtained from crude reaction mixture of zirconium containing species in a form of several colourless crystals. The molecular structure of this compound was determined by X-ray crystallography and revealed the presence of three LNN ligands in this complex (see Fig. 4). Similar structures are known from literature for –NHR R = tBu; 2,4,6-Me3C6H2 and 2,6-iPr2C6H3 ligands.18
In order to increase the stability of titanium compound 3 reactions of (η5-C5Me5)MCl3 (M = Ti, Zr, Hf) with LNNLi (2) were carried out. This ligand contains in addition to the NH group a lone electron pair on the tethered NMe2 group. The results show a different behaviour of the elements of group 4, which reflects their different capability of adopting higher coordination numbers. Thus, the titanium complex reacted with 2 under elimination of LiCl and afforded the complex (η5-C5Me5)LNNTiCl2 (7), where the dimethylamino group of the ligand is not involved into the bonding to the titanium centre (Scheme 2).
 | ||
| Scheme 2 Synthesis of compound 7. | ||
On the other hand zirconium and hafnium major products (η5-C5Me5)LNNMCl2 (M = Zr (8), Hf (9)) are stabilized via the intramolecular coordination of NMe2 group of the ligand. Half-sandwich zirconium trichloride reacts with one equivalent of 2 to form monomeric complex 8 in a high yield (Scheme 3).
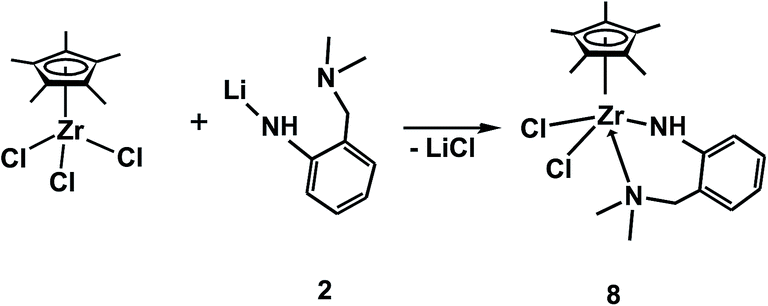 | ||
| Scheme 3 Synthesis of compound 8. | ||
Half-sandwich hafnium trichloride reacts with one equivalent of 2 in a similar way as the zirconium compound. As a minor product of this reaction several single-crystals of complex 10 were isolated from the crude mixture. The X-ray measurement determined the molecular structure of this product as the dimeric complex [(η5-C5Me5){µ-NC6H4(CH2(NMe2))}HfCl]2 (10), where two nitrogen atoms act as bridges between two hafnium atoms (Scheme 4). Formation of this compound is a consequence of the α-H abstraction from the LNN ligand by chloride atom with the assistance of a base, probably free LNNH molecule.
 | ||
| Scheme 4 Synthesis of compound 9. | ||
Solutions of complexes 8 and 9 in toluene-d8 exhibited a fluxional behaviour in the studied range of temperatures (228–328 K) as observed by NMR spectroscopy (see ESI†). At lower temperature the complexes possessed an unsymmetrical structure with the dimethylamino group coordinated to the metal centre (similarly as was proved in the solid state) which is in accordance with the presence of two singlets for non-equivalent methyl groups (2.17, 2.72 ppm for 8 and 2.15, 2.66 ppm for 9) and a system of two doublets for diastereotopic methylene hydrogens (2.51, 5.00 ppm, 2JHH = 13.3 Hz for 8 and 2.47, 4.93 ppm, 2JHH = 13.3 Hz for 9) in 1H NMR spectra. Upon heating, the signals became broader, coalesced and finally only an average signal could be observed as a consequence of fast interconversion between the pair of enantiomers (see Scheme 5).
 | ||
| Scheme 5 Dynamic behaviour of compounds 8 and 9 observed by NMR. | ||
The line-shape analysis with the WINDNMR program25 of spectra taken at different temperatures allowed us to determine the corresponding thermodynamic parameters (ΔH = 45.8 ± 1.4 kJ mol−1, ΔS = −24.3 ± 5.0 J K−1 mol−1, ΔG298 = 53.1 kJ mol−1 for 8; ΔH = 39.0 ± 1.2 kJ mol−1, ΔS = −37.4 ± 4.5 J K−1 mol−1, ΔG298 = 50.2 kJ mol−1 for 9) from the respective Eyring plots (see ESI†). The obtained ΔG values are lower in comparison to the corresponding LCN complexes (ΔG298 = 57.8 kJ mol−1 for Me5ZrLCN and ΔG298 = 64.6 kJ mol−1 for Me5HfLCN).23 We suggest that two phenomena contribute to the decrease of rotation barrier in LNN complexes. The higher electronic saturation of the metal with amide group (from LNN ligand) in comparison to aryl group (from LCN ligand) decrease the Lewis acidity of the metal and consequently weaken the coordination of the dimethylamine functionality as could be demonstrated with lower ΔH values in LNN when compared to LCN (ΔH = 77.4 ± 1.9 kJ mol−1 for Me5ZrLCN and ΔH = 66.9 ± 2.6 kJ mol−1 for Me5HfLCN).23 Furthermore, the formation of six-membered metal–amido–amino ring in LNN complexes (only 5-membered ring is constituted in LCN complexes) pushes the bulky aryl group away from the metal centre, which also facilitates its rotation leading to faster conformational exchange.
Infrared spectra of 8 and 9 complexes displayed similar absorption bands as previously described for compounds 4 and 5. Furthermore, stretching C–N vibrations were found for 8 and 9 in the range 1281–1246 cm−1.
Reactivity of LCN complexes towards lithium amides
Based on a successful Tinkertoy story by Lang26 who prepared multiheterometallic complexes, we followed a reverse approach performing thus reactions leading to the group 4 complexes containing four different ligands, which could be interesting from the point of view of mutual comparison of coordination abilities of selected donors and last but not least suggested formation of a stereogenic centre. Recently we reported the synthesis of group 4 metal complexes containing a pentamethyl- or tetramethyl-substituted cyclopentadienyl ring and C,N-chelating ligand LCN.23 In order to prepare new compounds containing four different ligands in coordination sphere of group 4 metals we have tested the reaction of zirconium and hafnium complexes of this type with lithium salts of 1 and 2. Titanium containing complexes were omitted due to lower stability of the corresponding amido compounds.(η5-C5Me4R)LCNMCl2 (M = Zr, Hf; R = Me, H) complexes react under mild conditions with 1 to eliminate LiCl together with LCN ligand and generate the dimeric products [(η5-C5Me4H){µ-NC6H3-2,6-iPr2}MCl]2 (M = Zr (11), Hf (13)) and [(η5-C5Me5){µ-NC6H3-2,6-iPr2}MCl]2 (M = Zr (12), Hf (14)) (Scheme 6). The likely intermediate containing bonded LN ligand is arising after transmetallation reaction and elimination of LiCl. In the next step the presence of amino group with reactive hydrogen atom in the neighbourhood of LCN ligand leads to the α-hydrogen abstraction and elimination of LCNH molecule. Finally the dimeric structure is formed. The driving force for the dimer formation is in this case the increase of electron density at the electron deficient central metal atom. Solid state structures of complexes 11–14 were determined by X-ray crystallography and the molecular structures were confirmed by NMR spectroscopy also in solutions. Both dimers have similar molecular structure as complex 10 obtained earlier as a minor side product as depicted in Scheme 4. Hafnium complex 14 was previously prepared by Tilley et al. by thermolysis of hafnium silyl complex Cp*Hf[Si(SiMe3)3]LNCl.8
 | ||
| Scheme 6 Preparation of compounds 11–14. | ||
1H and 13C NMR spectra of solutions of complexes 11, 12, 13, and previously published 14 differed from the above mentioned amide complexes 4 and 5. Apart from the missing NH signal, the most significant feature is the presence of two sets of doublets (centred at: 1.22 and 1.47 ppm in 12; 1.31 and 1.39 ppm in 11, 1.29, 1.51 ppm in 14,8 1.26 and 1.42 ppm in 13) for non-equivalent methyl groups of iso-propyl moiety. The spectral pattern is consistent with bridging imido moieties between two metal atoms, showing that the solid state structure (found by X-ray) is retained in the solution. Upfield shift of C5Me5 signal in 1H NMR spectra of imido complexes (δH C5Me5: 1.76 ppm in 12; 1.83 ppm in 14) in comparison to amide complexes 4 (δH C5Me5: 1.88 ppm) and 5 (δH C5Me5: 1.92 ppm) reflects higher electron saturation of the metal centre with two bridging imido ligands in comparison to chloride ligand.
Infrared spectra of these dimers 11–13 display aromatic C–H stretching vibration 3052 cm−1 and strong absorption for C–H out of plane deformation bands at 750 cm−1 typical for ortho-substituted benzene ligand. The asymmetric stretching vibration of methyl groups in Cp* and Cp′ ligands were observed at 2981–2869 cm−1 and the strong absorption band belonging to stretching C–N vibration was found at 1169 cm−1.
The results from the study of reactivity of LCN complexes with compound 1 encouraged us perform the same reactions with compound 2, too. We expected the formation of dimeric compounds with a bridging imide derived from the LNN ligand. Moreover the formation of the by-product 10 in previously mentioned reaction proved that such complexes can be obtainable.
The expectations were fulfilled only partially, results show differences in the behaviour of zirconium and hafnium derivatives, with a significant role of substitution on the cyclopentadienyl ring having an influence on the structure of the products.
Zirconium complexes containing permethylated and tetramethylated cyclopentadienyl ring (η5-C5Me4R)LCNZrCl2 (R = H, Me) were reacted with 2 under the same conditions as half-sandwich zirconium trichlorides (Schemes 7 and 8). The main products of the reactions were isolated as orange crystals suitable for X-ray analysis. Reaction mixtures as well as isolated crystals were studied by NMR and IR spectroscopy. In both cases, elimination of LiCl as a white precipitate and the liberated ligand (LCNH) was observed by NMR spectroscopy. The products are dimeric zirconium µ-imido complexes containing besides the bridging ligands two substituted cyclopentadienyl rings and two chlorine atoms [(η5-C5Me4H){µ-NC6H4-2-(CH2NMe2)-µ2N,N}ZrCl]2 (15) and [(η5-C5Me5){µ-NC6H4-2-(CH2NMe2)}ZrCl]2 (16).
 | ||
| Scheme 7 Synthesis of compound 15. | ||
 | ||
| Scheme 8 Synthesis of compound 16. | ||
While the tetramethylcyclopentadienyl-containing counterpart 15 shows a structure where pendant (dimethylamino)methyl arms are coordinated to the metal centres, the same N-donor groups in its Cp*-containing dimer 16 are uncoordinated to zirconium atoms.
NMR spectra of dimer 15 are consistent with the structure confirmed by X-ray diffraction (vide infra), which gives rise to one set of signals (syn-isomer) due to the C5Me4H and imide ligands. Coordinated pendant amine arms are likely undergoing dynamic conformational changes as evidenced by broad singlet signals for NMe2 groups. On the contrary, compound 16 was likely obtained as a mixture of syn-/anti-isomers from which only the crystals of the anti-isomer were analysed by X-ray diffraction (vide infra). NMR spectroscopy of the bulk crystalline product, however, reveals two sets of signals corresponding to both isomers. Both of them have the same spectral pattern with doublet signals due to diastereotopic methylene hydrogens of the LNN ligand, however, in comparison to 15 the difference in chemical shifts between these two signals is smaller and, furthermore, signals of NMe2 groups appear as sharp singlets. This all suggests that the (dimethylamino)methyl arms in both isomers of 16 remain uncoordinated in solution as well as in the solid state, which is probably caused by an increased steric bulkiness of the C5Me5 ligand.
Infrared spectra of complexes 15 and 16 displayed similar absorption bands as previously described for compounds 8 and 9 except the N–H stretching vibration which is missing.
In line with generally lower reactivity of Hf complexes (η5-C5Me4R)LNNLCNHfCl (R = H (17), Me (18)) were obtained from the reactions of complexes (η5-C5Me4R)LCNHfCl2 (R = Me, H) with 2 (Scheme 9). Both compounds were formed by a substitution of one chloride ligand with the corresponding LNN amide, while the LCN ligand remained unaffected. NMR spectra of both 17 and 18 showed the presence of both ligands as distinguishable sets of signals. The LNN amides gave rise to a characteristic NH signal in 1H spectra at δ 8.71 ppm for 17 or 8.46 ppm for 18, respectively. The presence of the NH group was confirmed by infrared spectra that show N–H stretching vibration at 3276 cm−1 for 17 and 3235 cm−1 for 18, respectively. The structure and stability of these monomeric hafnium complexes shows that the reaction pathway leading to formation of zirconium and hafnium dimers goes through such species. We can assume that the intermediate in the formation of dimeric complexes should be the same; however, in the case of zirconium being more prone to α-hydrogen abstraction that in the hafnium one.
 | ||
| Scheme 9 Preparation of compounds 17 and 18. | ||
Solid state study
Structures of 3–5 are the only crystallographically determined structures in the series reported in this paper, where the combination of one substituted Cp and one 2,6-(diisopropyl)phenylanilide (LN) is present in a mononuclear complex (for molecular structure of 5 see Fig. 1, for molecular structures of 3 and 4 see ESI†). Structures of 3–5 can be compared to the only example of such kind of compounds, containing both amide and Cp ligands – Cp*TiCl2NH-tBu,14 from the point of view of the structural arrangement, but the direct comparison of bond lengths and angles is possible only with 3, because of covalent radii differences between Ti and Zr/Hf atoms (∼0.15 Å). The ring centroid to the titanium atom distance is slightly (∼0.01 Å) longer in Cp*TiCl2NH-tBu14 than in 3, but a shortening of similar magnitude is found for the rest of the distances in the titanium atom vicinity. Mutual comparison of 3–5 reveals close structures with an elongation of metal–ligand distances for higher congeners of titanium only. On the other hand, a parallel between 5 and Cp*Hf(LN)3 (ref. 18) can be drawn, the Hf–Cg(Cp*) and all Hf–N are slightly elongated by ca. 0.06 Å in Cp*Hf(LN)3. The same trend can be observed in the case of angles Cg–Hf–N and C–N–Hf which are in Cp*Hf(LN)3 more opened by ca. 5 and 20°, respectively, probably because of high steric repulsion of three anilide ligands. | ||
| Fig. 1 Molecular structure of 5 (ORTEP view, 30% probability level). Hydrogen atoms are omitted (except of H1) for clarity. Selected interatomic distances [Å] and angles [°]: Hf1–N1 2.003(4), Hf1–Cl1 2.3699(17), Hf1–Cl2 2.3680(16), Hf1–Cg1 2.144(3), Cg1–Hf1–Cl1 113.2(4), Cg1–Hf1–Cl2 115.9(3), Cg1–Hf1–N1 111.5(3), C1–N1–Hf1 121.4(3). | ||
In the series where the combination of one substituted Cp and one potentially chelating ligand 2-(dimethylaminomethyl)anilide (LNN) are present, two rather different structures were determined by X-ray diffraction techniques. The first one containing titanium 7 (Fig. 2) is very close to the classical piano stool structure of Cp*TiCl2NH-tBu14a with only Ti–N bond slightly elongated due to the presence of N–H⋯N intramolecular connection. N3 atom of the flexible, potentially chelating arm is out of the primary coordination sphere of the titanium atom as well as from the parent phenyl ring (55.5(4)°).
 | ||
| Fig. 2 Molecular structure of 7 (ORTEP view, 50% probability level). Hydrogen atoms (except of H1) and the second independent molecule are omitted for clarity. Selected interatomic distances [Å] and angles [°] (appropriate parameters for the second independent molecule are given in parenthesis): Ti1–N1 1.906(3) (1.912(2)), Ti1–N3 4.691(3) (4.637(3)), Ti1–2.2775(10) (2.2783(10)), Ti1–Cl2 2.2648(10) (2.2601(10)), Ti–Cg 2.041(3) (2.028(3)), N1–H1⋯N3 2.978(4) (2.922(3)); Cg–Ti1–Cl1 118.1(3) (117.4(3)), Cg–Ti1–Cl2 116.7(4) (115.5(3)), Cg–T1–N1 110.1(3) (110.7(3)), C1–N1–Ti1 130.7(2) (130.0(2)), C3–C2–C7–N3 55.5(4). | ||
In the structure of the second representative 8 (Fig. 3, structures of two geometrically similar solvato-polymorphs of 8 (8 and 8′) were determined), this arm is closely interacting with the zirconium center forming nearly perfect trigonal bipyramid. One can estimate, this particular interaction is stronger than the suggested hydrogen bond between two nitrogen atoms found in 7. The axial positions are occupied by the coordinated nitrogen atom N2 and the centroid of the Cp* ring, while the Zr1–N2 distance of 2.513(3) Å is in the range of the medium strong interactions of these atoms taking into the account the sum of covalent and van der Waals radii of both atoms.27 Similar distances were also found for interactions of the same type of atoms in complexes of Cp-substituted zirconium complexes bearing related C,N-chelating ligand where the coordination polyhedra of the zirconium atom is being tetragonal pyramid.23,28 The equatorial plane is a bit distorted towards the N2 atom, when a plane is defined by two chlorine and nitrogen N1 atoms, the Zr1 atom is 0.509 Å above this plane which is nearly parallel to the plane of Cp* ring.
 | ||
| Fig. 3 Molecular structure of 8 toluene (ORTEP view, 40% probability level). Hydrogen atoms (except of H1) and toluene solvent molecule are omitted for clarity. Selected interatomic distances [Å] and angles [°]: Zr1–N1 2.093(3), Zr1–N2 2.513(3), Zr1–Cl1 2.4310(10), Zr1–Cl2 2.4511(10), Zr1–Cg1 2.245(4), Cg1–Zr1–Cl1 104.5(3), Cg1–Zr1–Cl2 103.1(2), Cg1–Zr1–N1 100.9(3), Cg1–Zr1–N2 175.2(3), C1–N1–Zr1 140.8(3), C3–C2–C7–41.8(3). | ||
Structure of 6 (Fig. 4), as a minor byproduct isolated from preparation of 4 is closely related to the reported structures of Cp*Zr(NH-tBu)3 and Cp*Hf(LN)3, respectively.18 High steric repulsion of three LN substituted ligands in comparison to ligands with t-Bu substituents in Cp*Zr(NH-tBu)3 is obviously a reason for different arrangement of ligands in all these three compounds (a syn for tBu and an alternate orientation of amido ligands in LN substituted complexes). In a comparison of relevant geometrical parameters of 6, to appropriate ones of 4 and 8, slightly longer distances in the primary coordination sphere of zirconium atom were observed.
 | ||
| Fig. 4 Molecular structure of 6 (ORTEP view, 30% probability level). Hydrogen atoms are omitted (except of H1, H2 and H3) for clarity. Selected interatomic distances [Å] and angles [°]: Zr1–N1 2.0740(16), Zr1–N2 2.0642(16), Zr1–N3 2.0760(15), Zr1–Cg1 2.255(4); Cg1–Zr1–N1 111.45(3), Cg1–Zr1–N2 115.18(3), Cg1–Zr1 115.30(3). | ||
Seven structures of dimers (dinuclear complexes) were determined. All four combinations of Zr, Hf, Cp′ and Cp* 2,6-(diisopropyl)phenyl-substituted imido ligand complexes 11–14 are from a structural point of view closely related, having central binuclear character, bridged by two imides, with nearly square planar M2N2 ring structures and M − N bonds being only slightly unequal. The shape of the metals' coordination polyhedra is pseudotetrahedral. The parallel orientation of the same type of ligand rings, both Cp and phenyls, and metal–chlorine atom bonds with an anti-orientation with respect to the plane or the central M2N2 motif, is the general phenomenon observed in all these structures. In zirconium 11 (Fig. 5) and 12 (Fig. 6) as well as in hafnium 13 (Fig. 7) and 14 (for molecular structure see ESI Fig. S3†) compounds, the interatomic distances are slightly longer for Cp* substituted complex probably due to higher sterical hindrance and electron density of this substituent. The metal–metal distances being around 3.05 Å in all complexes.
 | ||
| Fig. 5 Molecular structure of 11 (ORTEP view, 50% probability level, symmetry code: (a) −x, −y + 1, −z + 1). Hydrogen atoms are omitted for clarity. Selected interatomic distances [Å] and angles [°]: Zr1–N1 2.1530(16), Zr1–N1a 2.0277(16), Zr1–Cl1 2.4153(5), Zr1–Cg1 2.219(3), Zr1–Zr1a 3.064(4), Cg1–Zr1–Cl1 108.18(3), Cg1–Zr1–N1 122.69(3), Cg1–Zr1–N1a 124.05(2), Cg–Zr1–Zr1a 138.59(3), N1–Zr1–N1a 85.78(6), N1–Zr1–Cl1 106.25(4), N1a–Zr1–Cl1 107.22(5), Zr1–N1–Zr1a 94.22(6), C10–N1–Zr1 111.79(11), C10–N1–Zr1a 153.06(13). | ||
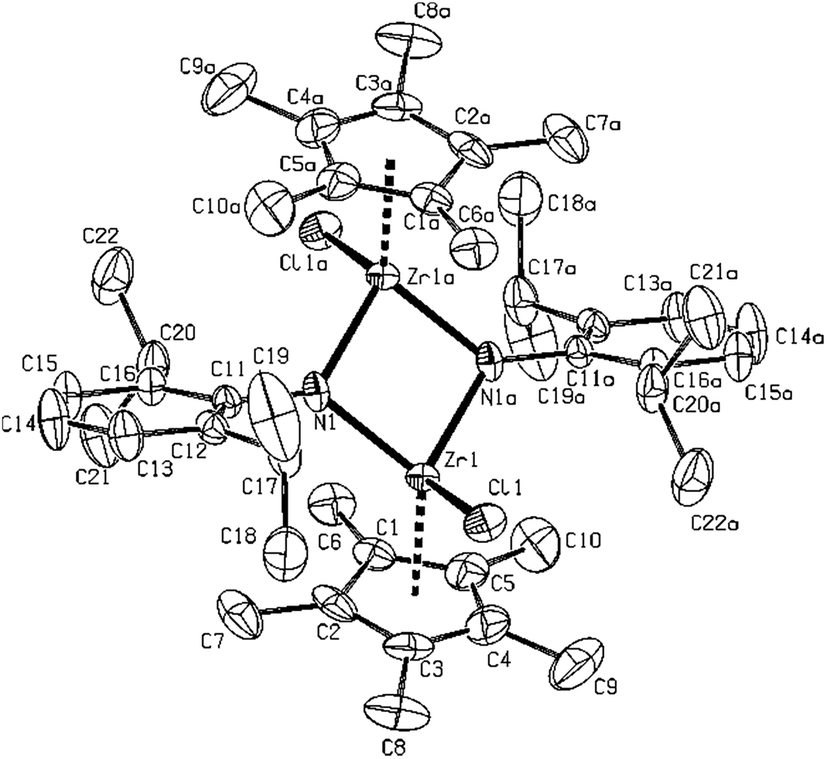 | ||
| Fig. 6 Molecular structure of 12 (ORTEP view, 30% probability level, symmetry code: (a) −x + 1/2, −y + 1/2, −z + 1). Hydrogen atoms are omitted for clarity. Selected interatomic distances [Å] and angles [°]: Zr1–N1 2.086(4), Zr1–N1a 2.106(4), Zr1–Cl1 2.4280(14), Zr1–Cg1 2.234(3), Zr1–Zr1a 3.092(4), Cg1–Zr1–Cl1 106.42(3), Cg1–Zr1–N1 122.69(2), Cg1–Zr1–N1a 123.79(3), Cg–Zr1–Zr1a 138.00(3), N1–Zr1–N1a 84.96(14), N1–Zr1–Cl1 109.17(11), N1a–Zr1–107.94(11), Zr1–N1–Zr1a 95.05(14), C11–N1–Zr1 135.5(3), C11–N1–Zr1a 129.5(3). | ||
 | ||
| Fig. 7 Molecular structure 13 (ORTEP view, 50% probability level, symmetry code: (a) −x + 1, −y, −z + 1). Hydrogen atoms are omitted for clarity. Selected interatomic distances [Å] and angles [°]: Hf1–N1 2.026(3), Hf1–N1a 2.115(3), Hf1–Cl1 2.3918(9), Hf1–Cg1 2.193(4), Hf1–Hf1a 3.0373(3), N1–Hf1–N1a 85.65(11), N1–Hf1–Cl1 107.14(8), N1a–Hf1–Cl1 105.14(8), Cl1–Hf1–Cg1 108.64(9), N1–Hf1–Cg1 125.23(8), N1a–Hf1–Cg1 122.00(9), N1–Hf1–Hf1a 43.97(8), N1a–Hf1–Hf1a 41.68(8), C16–Hf1–Hf1a 164.22(9). | ||
The structure of the analogue of 11–13, compound 14, has been determined earlier by Tilley,8 but the dataset quality did not allowed an anisotropic refinement of lighter atoms. We re-determined the structure in order to have the exact comparison in hands. Again, the hafnium complexes 13 and 14 have slightly shorter metal to other element distances than the zirconium compounds 11 and 12 because of different covalent radii of both atoms while the values of relevant interatomic angles for 11–14 are comparable.
Another three structures of complexes 10, 15 and 16 bearing the potentially N,N-chelating ligand were determined. Mutual discrepancy in Hf–N bond lengths within the Hf2N2 core of 10 (Fig. 8) is a bit larger than in previous cases of 11–14, probably because of lower bulkiness of the imido ligand but the rest of the parameters is quite similar. In this respect, the Cp* zirconium complex 16 (Fig. 9) reveals close structures to 11–14 and 10 with the same arrangement and mutual orientation of the ligands. On the other hand, the zirconium–zirconium distance in 16 is elongated by ∼0.06 Å in comparison to the rest of discussed dinuclear complexes. In this respect, the structure of complex 15 (Fig. 10), bearing the chelating LNN and Cp′ ligands is also dinuclear but the orientation of Cp rings and chloro ligands is mutually syn. The aromatic rings are perpendicularly oriented with pendant amino groups being as strongly coordinated to the zirconium atoms as in the case of 4. The Zr–Cl and Zr–Cg1, and more significantly in the Zr1–Zr1a case, these distances are longer than in the rest of dinuclear cases, because of an increase of electron density on the zirconium atoms, and also by the change of the pseudotetrahedral metal neighborhood to the square pyramidal one.
 | ||
| Fig. 8 Molecular structure of 10 (ORTEP view, 50% probability level, symmetry code: (a) −x, −y, −z + 2). Hydrogen atoms are omitted for clarity. Selected interatomic distances [Å] and angles [°]: Hf1–N1 2.012(3), Hf1–N1a 2.106(3), Hf1–Cl1 2.4090(8), Hf–N2 5.172(3), Hf1–Cg1 2.174(3), Cg1–Hf1–Cl1 111.74(3), Hf1–Hf1a 3.068(4), Cg1–Hf1–N1 117.55(3), Cg1–Hf1–N1a 123.55(3), Cg1–Hf1–Hf1a 133.06(3), N1–Hf1–N1a 83.71(11), N1–Hf1–Cl1 107.63(8), N1a–Hf1–Cl1 109.13(8), Hf1–N1–Hf1a 96.29(11), C1–N1–Hf1 147.4(2), C1–N1–Hf1a 115.6(2), C3–C2–C7–N2 11.75(3). | ||
 | ||
| Fig. 9 Molecular structure of 16 (ORTEP view, 50% probability level, symmetry code: (a) −x, −y + 1, −z + 1). Hydrogen atoms are omitted (except of H1) for clarity. Selected interatomic distances [Å] and angles [°]: Zr1–N1 2.0024(19), Zr1–N1a 2.1427(19), Zr1–Cl1 2.4471(6), Zr1–Zr1a 3.1045(5), Cg1–Zr1 2.200(6), N1–Zr1–N1a 83.06(8), N1–Zr1–Cl1 105.72(5), N1–Zr1–Cl1 112.97(5), N1–Zr1–Zr1a 43.25(5), N1a–Zr1–Zr1a 39.81(5), Cl1–Zr1–Zr1a 116.370(18), Cg–Zr1–Zr1a 131.90(11), Cg–Zr1–Cl1 111.02(12). | ||
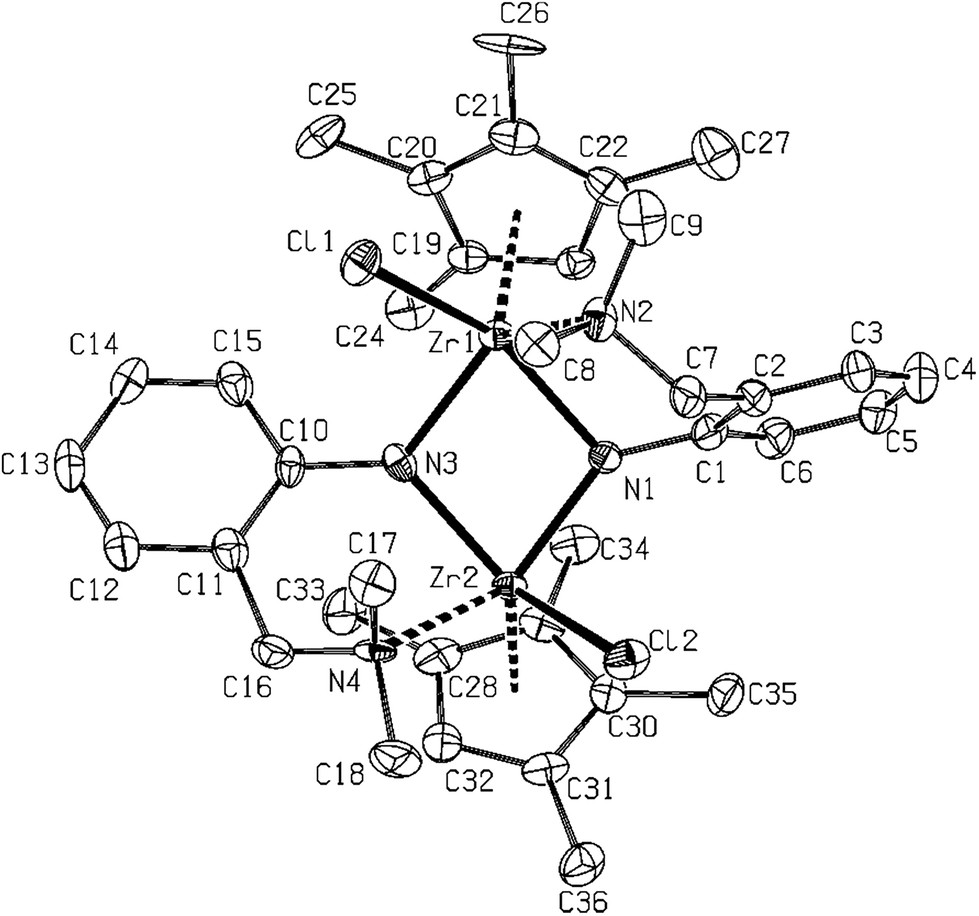 | ||
| Fig. 10 Molecular structure of 15 (ORTEP view, 50% probability level). Hydrogen atoms are omitted for clarity. Selected interatomic distances [Å] and angles [°]: Zr1–N3 2.090(7), Zr1–N1 2.111(7), Zr1–Cl1 2.512(2), Zr1–N2 2.569(7), Zr1–Zr2 3.2352(12), Zr2–N1 2.060(7), Zr2–N3 2.123(7), Zr2–N4 2.447(7), Cg1–Zr1 2.297(5), Cg2–Zr2 2.259(5), N3–Zr1–N1 78.2(3), N3–Zr1–Cl1 88.7(2), N1–Zr1–Cl1 143.69(19), N3–Zr1–N2 120.9(3), N1–Zr1–N2 79.4(3), Cl1–Zr1–N2 78.74(17), Cg1–Zr1–Cl1 103.22(12), Cg2–Zr2–Cl2 106.92(11), Cg1–Zr1–Zr2 131.28(9), Cg2–Zr2–Zr1 134.58(13), C3–C2–C7–N2 62.21(13), C12–C11–C16–N4 19.25(13). | ||
 | ||
| Fig. 11 Molecular structure of 17 (ORTEP view, 50% probability level). Hydrogen atoms are omitted (except of H2) for clarity. Selected interatomic distances [Å] and angles [°]: Hf1–N2 2.055(4), Hf1–C1 2.276(5), Hf1–Cl1 2.4440(13), Hf1–N1 2.463(4), Cg1–Hf1 2.194(4), N2–Hf1–C1 90.01(17), N2–Hf1–Cl1 91.14(11), C1–Hf1–Cl1 133.62(12), N2–Hf1–N1 141.65(15), C1–Hf1–N1 70.87(15), Cl1–Hf1–N1 80.15(10), Cg1–Hf1–Cl1 115.19(12), Cg1–Hf1–N2 106.15(11), C3–C2–C7–N1 30.31(9), N2–H2⋯N3 2.960(5). | ||
The last complex under structural investigation 17 is the compound containing four different ligands – Cp′, N,N′-chelating (LNN), C,N-chelating (LCN) and chloro ones, accommodated in the coordination polyhedra of the central hafnium atom (Fig. 11). Only the LCN ligand is doubly coordinated/covalently bound to the metal with the metal to carbon and nitrogen distances within the range of sum of van der Waals and covalent radii, respectively. The amido function from the potentially N,N′-chelating ligand, both chelators from the C,N-chelating ligand as well as the chloro ligand are located within the basal plane of the deformed square pyramid, while the fifth substituent is the Cp′ ligand on the top of it. All the distances are slightly longer than in both studied mono- and dinuclear complexes but comparable to the sterically hindered complex Cp*Hf(LN)3.18
Experimental
General experimental
All operations were carried out under an argon atmosphere using standard Schlenk techniques. 1H (300.0 MHz), 13C{1H} (75.4 MHz) NMR spectra were recorded on a Varian Mercury 300 spectrometer at 298 K, if not otherwise stated. Some 1H (500.13 MHz) NMR spectra were recorded on a Bruker Avance III 500 spectrometer, equipped with a z-gradient 5 mm Prodigy™ cryoprobe. Chemical shifts (δ/ppm) are given relative to solvent signals C6D6: δH 7.15 ppm, δC 128.00 ppm; toluene-d8: δH (CH2D signal) 2.08 ppm, δC (CD3 signal) 20.43 ppm; CDCl3: δH 7.26 ppm, δC 77.16 ppm; CD2Cl2: δH 5.32 ppm, δC 53.84 ppm, C6D5Br: δH 7.30 ppm, δC 122.54 ppm. EI-MS spectra were measured on a VG-7070E mass spectrometer at 70 eV. IR spectra of samples in KBr pellets were measured in an air-protecting cuvette on a Nicolet Avatar FT IR spectrometer in the range 400–4000 cm−1. KBr pellets were prepared in a glovebox Labmaster 130 (mBraun) under purified nitrogen. Elemental analyses were carried out on a FLASH EA1112 CHN/O Automatic Elemental Analyzer (Thermo Scientific).Chemicals
The solvents THF, hexane, and toluene were dried by distillation from sodium/benzophenone and stored over sodium mirror (η5-C5Me5)MCl3 (M = Ti, Zr, Hf) were purchased from Sigma Aldrich and used directly as obtained. (η5-C5Me4R)LCNMCl2 (M = Zr, Hf; R = Me, H; LCN = C6H4-2-(CH2NMe2)-µ2-C,N) were synthesized according to a published procedure.23 Lithium salts of 2,6-diisopropylaniline Li[NHC6H3-2,6-(CHMe2)2] (LNLi) and 2-[(dimethylamino)methyl]aniline Li[NHC6H4-2-(CH2NMe2)] (LNNLi) were obtained by published methods.29X-ray crystallography
The X-ray data obtained from crystals for all compounds were acquired at 150 K using an Oxford Cryostream low-temperature device on a Nonius KappaCCD diffractometer with Mo Kα radiation (λ = 0.71073 Å), a graphite monochromator, and in the Φ and χ scan mode. Data reductions were performed with DENZO-SMN.30 The absorption was corrected by integration methods.31 Structures were solved by direct methods (Sir92)32 and refined by full matrix least-square based on F2 (SHELXL97).33 Hydrogen atoms were mostly localized on a difference Fourier map, but in order to ensure uniformity of the treatment of the crystal, all hydrogen atoms were recalculated into idealized positions (riding model) and assigned temperature factors Hiso (H) = 1.2 Ueq (pivot atom) or of 1.5 Ueq for the methyl moiety with C–H = 0.96, 0.97, 0.98 and 0.93 Å for methyl, methylene, methine and hydrogen atoms in aromatic rings, or multiply bounded C–H groups respectively. Some of the hydrogen atoms attached to N atoms were located on the Fourier difference map while the other ones were added with bond length of 0.89 Å. Structures of 3, 4, 5, 8′ are disordered and the disorder was treated by standard procedures of SHELXL program splitting some atoms into two parts.There are solvent accessible voids within the crystals of 8. The PLATON/SQUEZZE34 procedures were applied in order to correct these voids and the presence of one molecule of toluene has been evaluated within the unit cell.
Synthesis of compounds
LNLi (1). 1H NMR (500.13 MHz, 295 K, THF-d8) δ: 1.19 (d, 3JHH = 6.8 Hz, 12H, Me), 2.70 (br s, 1H, NH), 3.19 (br s, 2H, CH), 6.05 (br s, 1H, ArH), 6.68 (br s, 2H, ArH).
LNNLi (2). 1H NMR (500.13 MHz, 295 K, C6D6) δ: 1.70 (s, 6H, Me2), 2.73 (s, 1H, NH), 2.97 (br s, 2H, CH2), 6.48 (t, 3JHH = 7.2 Hz, 1H, ArH), 6.60 (d, 3JHH = 7.1 Hz, 1H, ArH), 6.79 (d, 3JHH = 6.8 Hz, 1H, ArH), 7.13 (t, 3JHH = 7.2 Hz, 1H, ArH).
(η5-C5Me5){NHC6H3-2,6-iPr2}TiCl2 (3). Yellow crystals. 1H NMR (300 MHz, toluene-d8) δ: 1.26 (d, 3JHH = 6.6 Hz, 12H, CHMe2), 2.07 (s, 15H, C5Me5), 3.68 (septet, 3JHH = 6.6 Hz, 2H, CHMe2), 6.90–7.10 (m, 3H, C6H3), 9.51 (br s, 1H, NH). 13C{1H} NMR (toluene-d8) δ: 13.3 (C5Me5), 24.0 (CHMe2), 28.7 (CHMe2), 124.2, 126.4 (2 × C6H3 CH), 133.8 (C5Me5), 137.1 (C–CHMe2), 140.3 (C–NH).
(η5-C5Me5){NHC6H3-2,6-iPr2}ZrCl2 (4). Light orange crystals, yield 0.8 g (56%). 1H NMR (300 MHz, toluene-d8) δ: 1.20 (d, 3JHH = 6.8 Hz, 12H, CHMe2), 1.88 (s, 15H, C5Me5), 3.23 (septet, 3JHH = 6.8 Hz, 2H, CHMe2), 6.37 (br s, 1H, NH), 7.01–7.10 (m, 3H, C6H3). 13C{1H} NMR (toluene-d8) δ: 11.7 (C5Me5), 23.4 (CHMe2), 29.7 (CHMe2), 123.0 (C6H3), 124.8 (C5Me5), 138.6 (C–CHMe2), 147.5 (C–NH). IR (KBr, cm−1): 3352 (w), 3061 (w), 2961 (vs), 2920 (s), 2869 (s), 1622 (m), 1460 (s), 1430 (s), 1380 (m), 1360 (m), 1322 (m), 1263 (m), 1176 (m), 1113 (w), 1094 (w), 1057 (m), 1044 (w), 1025 (m), 930 (w), 887 (w), 836 (w), 799 (m), 750 (s), 687 (m), 634 (m), 609 (m), 590 (m), 531 (m), 470 (w), 444 (w). M.p. = 120–125 °C. Anal. calc. for C22H33NCl2Zr: C, 55.79; H, 7.02; N, 2.96. Found: C, 55.71; H, 6.98; N, 2.93.
(η5-C5Me5){NHC6H3-2,6-iPr2}HfCl2 (5). Pale yellow crystals, yield 1.1 g (65%). 1H NMR (300 MHz, toluene-d8) δ: 1.20 (d, 3JHH = 6.8 Hz, 12H, CHMe2), 1.92 (s, 15H, C5Me5), 3.30 (septet, 3JHH = 6.8 Hz, 2H, CHMe2), 6.02 (br s, 1H, NH), 6.96–7.08 (m, 3H, C6H3). 13C{1H} NMR (toluene-d8) δ: 11.4 (C5Me5), 23.5 (CHMe2), 29.3 (CHMe2), 123.0 (C5Me5), 123.2 (m – CH, C6H3), 125.6 (p – CH, C6H3), 142.7 (C–NH), 143.4 (C–CHMe2). IR (KBr, cm−1): 3355 (w), 3062 (w), 2962 (vs), 2922 (s), 2870 (s), 1622 (m), 1437 (s), 1460 (s), 1381 (s), 1360 (m), 1321 (m), 1263 (m), 1220 (m), 1181 (m), 1113 (m), 1094 (w), 1057 (w), 1044 (w), 1027 (w), 957 (w), 929 (w), 886 (w), 841 (m), 799 (s), 750 (vs), 689 (w), 649 (w), 622 (m), 533 (w), 495 (w), 444 (w). M.p. = 142–144 °C. Anal. calc. for C22H33NCl2Hf: C, 47.11; H, 5.93; N, 2.50. Found: C, 47.05; H, 5.89; N, 2.44.
(η5-C5Me5){NHC6H4-2-(CH2NMe2)}3Zr (6). Only several crystals of compound 6 were isolated as a side product during preparation of 4. For crystal structure of 6 see Fig. 4. Similar structures are known for –NHR R = tBu; 2,4,6-Me3C6H2 and 2,6-iPr2C6H3 ligands.18
(η5-C5Me5){NHC6H4-2-(CH2NMe2)}TiCl2 (7). Red crystals, yield 0.67 g (55%). 1H NMR (300 MHz, CDCl3) δ: 2.28 (s, 15H, C5Me5), 2.35 (s, 6H, NMe2), 3.56 (s, 2H, CH2NMe2), 6.76–6.82, 6.93–6.97, 7.24–7.30, 7.77–7.80 (4 × m, 4 × 1H, CH, C6H4), 12.45 (br s, 1H, NH). 13C{1H} NMR (CDCl3) δ: 13.2 (C5Me5), 44.8 (NMe2), 63.7 (CH2NMe2), 119.0 (C–CH2, C6H4), 122.1, 123.6, 129.1 (3 × CH, C6H4), 129.4 (C5Me5), 129.6 (CH, C6H4), 154.4 (C–N, C6H4). IR (KBr, cm−1): 2958 (m), 2912 (m), 2857 (m), 2822 (m), 2776 (m), 1589 (m), 1573 (w), 1553 (w), 1485 (s), 1467 (s), 1380 (m), 1316 (s), 1249 (s), 1229 (m), 1177 (w), 1153 (w), 1099 (m), 1022 (m), 962 (m), 936 (w), 885 (w), 864 (w), 845 (w), 796 (m), 759 (s), 721 (w), 661 (w), 621 (w), 537 (w), 520 (w), 472 (w), 450 (m). M.p. = 152 °C. Anal. calc. for C19H28N2Cl2Ti: C, 56.60; H, 7.00; N, 6.95. Found: C, 56.54; H, 6.95; N, 6.91.
(η5-C5Me5){NHC6H4-2-(CH2NMe2)-µ2-N,N}ZrCl2 (8). Yellow crystals, yield 0.87 g (65%). 1H NMR (300 MHz, 238 K, toluene-d8) δ: 1.91 (s, 15H, C5Me5), 2.17 (s, 3H, NMe2), 2.51 (d, 1H, 2JHH = 13.3 Hz, CH2NMe2), 2.72 (s, 3H, NMe2), 5.00 (d, 1H, 2JHH = 13.3 Hz, CH2NMe2), 6.15–6.23 (m, 1H, CH, C6H4), 6.51–6.61 (m, 2H, CH, C6H4), 6.82–7.01 (m, 1H, CH, C6H4), 7.34 (br s, 1H, NH). 13C{1H} NMR (238 K, toluene-d8) δ: 12.2 (C5Me5), 46.5, 51.3 (2 × NMe2), 64.0 (CH2NMe2), 116.7, 119.4, 123.8, 129.4, 130.5 (CH and C–CH2 and C6H4), 123.4 (C5Me5), 150.6 (C6H4, C–N). IR (KBr, cm−1): 3376 (m), 3023 (m), 2985 (m),2908 (s), 2866 (m), 1936 (w), 1897 (w), 1859 (w), 1824 (w),1786 (w),1598 (s), 1585 (s), 1495 (s), 1476 (m), 1455 (s), 1428 (s), 1403 (m), 1379 (s), 1358 (m), 1313 (s),1281 (s),1246 (s), 1192 (m),1175 (w),1158 (w),1145 (w),1108 (m),1067 (w),1048 (m),1020 (m), 993 (m), 965 (w), 931 (w), 889 (m),858 (w), 838 (m), 798 (m), 755 (s), 721 (m), 696 (w), 640 (m), 614 (m), 544 (m), 488 (w),452 (w),410 (w). M.p. = 150 °C. Anal. calc. for C19H28N2Cl2Zr: C, 51.10; H, 6.32; N, 6.27. Found: C, 51.01; H, 6.22; N, 6.21.
(η5-C5Me5){NHC6H4-2-(CH2NMe2)-µ2-N,N}HfCl2 (9). Yellow crystals, yield 1.09 g (68%). 1H NMR (300 MHz, 228 K, toluene-d8) δ: 1.95 (s, 15H, C5Me5), 2.15, 2.66 (2 × s, 3H, NMe2), 2.47, 4.93 (2 × d, 1H, 2JHH = 13.3 Hz, CH2NMe2), 6.26–6.38 (m, 1H, CH, C6H4), 6.45 (br s, 1H, NH), 6.58–6.72 (m, 2H, CH, C6H4), 7.04–7.14 (m, 1H, CH, C6H4). 13C{1H} NMR (228 K, toluene-d8) δ: 12.3 (C5Me5), 46.7, 51.4 (2 × NMe2), 63.8 (CH2NMe2), 117.8, 119.1, 121.2, 123.5, 129.9 (CH and C–CH2 and C6H4), signal C5Me5 is overlapped by solvent signal, 150.4 (C6H4, C–N). IR (KBr, cm−1): 3270 (w),2858 (s), 2816 (m), 2773 (m), 1600 (s), 1486 (s), 1458 (s), 1427 (m), 1401 (w), 1378 (m), 1363 (m), 1315 (m), 1272 (s), 1253 (s), 1179 (w), 1148 (w), 1108 (m), 1044 (w), 1018 (m), 985 (w), 927 (w), 891 (m), 832 (w), 800 (m), 752 (s), 730 (m), 695 (m), 640 (w), 608 (w), 591 (w), 546 (w), 488 (w), 466 (w). M.p. = 150 °C. Anal. calc. for C19H28N2Cl2Hf: C, 42.75; H, 5.29; N, 5.25. Found: C, 42.71; H, 5.22; N, 5.19.
[(η5-C5Me5){µ-NC6H4-2-(CH2NMe2)}HfCl]2 (10). Only several crystals of compound 10 were isolated as a side product during preparation of 9. No other analyses could have been performed due to low amount and decomposition of the sample during X-ray analysis. For crystal structure of 10 see Fig. 8.
[(η5-C5Me4H){µ-NC6H3-2,6-iPr2}ZrCl]2 (11). Light orange crystals, yield 1.95 g (77%). 1H NMR (300 MHz, C6D6) δ: 1.31, 1.39 (2 × d, 3JHH = 6.5 Hz, 12H, CHMe2), 1.57, 2.13 (2 × s, 12H, C5Me4), 4.24 (septet, 3JHH = 6.7 Hz, 4H, CHMe2), 5.72 (s, 2H, HC5Me4), 7.02–7.13 (m, 6H, C6H3). 13C{1H} NMR (C6D6) δ: 10.9, 13.1 (2 × C5Me4), 25.8, 28.1 (2 × CHMe2), 28.5 (CHMe2), 113.5 (CH, C5Me4H), 124.3 (CH, C6H3), 125.4, 125.4 (2 × Cipso, C5Me4H), 144.1 (C–CHMe2, C6H3), 145.5 (C–N, C6H3). IR (KBr, cm−1): 3052 (m), 2973 (s), 2938 (s), 2915 (s), 2869 (m), 2729 (w), 1914 (w), 1623 (w), 1585 (w), 1505 (w), 1495 (m), 1412 (s), 1359 (m), 1384 (m), 1317 (m), 1238 (m), 1144 (m), 1169 (vs), 1109 (s), 1097 (m), 1042 (w), 985 (w), 926 (w), 887 (s), 863 (s), 818 (s), 797 (m), 750 (vs), 707 (m), 631 (w), 613 (w), 588 (w), 556 (s), 483 (s), 452 (w), 406 (m). M.p. = 243 °C. Anal. calc. for C42H60N2Cl2Zr2: C, 59.61; H, 7.15; N, 3.31. Found: C, 59.53; H, 7.08; N, 3.29.
[(η5-C5Me5){µ-NC6H3-2,6-iPr2}ZrCl]2 (12). Light orange crystals, yield 1.86 g (71%). 1H-NMR (300 MHz, toluene-d8) δ: 1.22, 1.47 (2 × d, 3JHH = 6.4 Hz, 12H, CHMe2), 1.76 (s, 30H, C5Me5), 4.25 (septet, 3JHH = 6.4 Hz, 4H, CHMe2), 6.94 (t, 3JHH = 7.5 Hz, 2H, C6H3), 7.06 (d, 3JHH = 7.5 Hz, 4H, C6H3). 13C{1H} NMR (toluene-d8) δ: 11.7 (C5Me5), 25.4, 28.5 (2 × CHMe2), 30.4 (CHMe2), 124.0 (C5Me5), 124.7 (CH, C6H3), 141.6 (C–CHMe2, C6H3), 148.2 (C–N, C6H3). IR (KBr, cm−1) 3052 (w), 2978 (s), 2953 (s), 2915 (s), 2871 (m), 1588 (w), 1458 (m), 1413 (m), 1381 (m), 1360 (w), 1318 (w), 1294 (w), 1250 (w), 1228 (w), 1162 (s), 1143 (s), 1100 (s), 1039 (w), 1025 (w), 926 (w), 885 (m), 854 (m), 797 (w), 751 (s), 705 (w), 624 (w), 611 (w), 599 (w), 556 (s), 482 (s), 414 (m). M.p. = 240 °C. Anal. calc. for C44H64N2Cl2Zr2: C, 60.44; H, 7.38; N, 3.20. Found: C, 60.38; H, 7.31; N, 3.16.
[(η5-C5Me4H){µ-NC6H3-2,6-iPr2}HfCl]2 (13). Yellow crystals, yield 2.50 g (82%). 1H NMR (300 MHz, toluene-d8) δ: 1.26, 1.42 (2 × d, 3JHH = 6.6 Hz, 12H, CHMe2), 2.04, 2.19 (2 × s, 12H, C5Me4), 4.35 (septet, 3JHH = 6.6 Hz, 4H, CHMe2), 5.72 (s, 2H, HC5Me4), 6.86–6.92 (m, 2H, C6H3), 7.07–7.11 (m, 4H, C6H3). 13C{1H} NMR (toluene-d8) δ: 10.9, 13.1 (2 × C5Me4), 26.1 (CHMe2), 28.0 (CHMe2), 29.0 (CHMe2), 112.5 (CH, C5Me4H), 122.6, 122.9 (2 × Cipso, C5Me4H), 124.2, 128.2 (2 × CH, C6H3), 143.0 (C–CHMe2), 146.0 (C–N, C6H3). IR (KBr, cm−1) 3053 (w), 2981 (m), 2968 (m), 2949 (m), 2938 (m), 2915 (m), 2869 (w), 1460 (w), 1414 (m), 1384 (w), 1359 (w), 1318 (w), 1296 (w), 1237 (w), 1169 (s), 1144 (w), 1108 (m), 1097 (w), 1037 (w), 887 (m), 867 (w), 821 (m), 798 (w), 751 (s), 708 (w), 612 (w), 568 (s), 496 (m), 452 (w), 407 (w). M.p. = 263 °C. Anal. calc. for C42H60N2Cl2Hf2: C, 49.42; H, 5.92; N, 2.74. Found: C, 49.32; H, 5.84; N, 2.71.
[(η5-C5Me5){µ-NC6H3-2,6-iPr2}HfCl]2 (14). Synthesized according to procedures published elsewhere.8 Yellow crystals, yield 2.23 g (71%).
[(η5-C5Me4H){µ-NC6H4-2-(CH2NMe2)-µ2-N,N}ZrCl]2 (15). Orange crystals; yield 0.18 g (45%). 1H NMR (300 MHz, C6D5Br) δ: 1.48, 1.72, 2.15, 2.28 (4 × s, 3H, C5Me4), 2.32 (br s, 6H, NMe2), 3.12, 3.95 (2 × d, 2JHH = 13.5 Hz, 1H, CH2), 5.80 (s, 1H, C5Me4H), 6.51–7.37 (m, 4H, C6H4). 13C{1H} NMR(C6D5Br) δ: 11.8, 13.0, 13.7, 14.6 (4 × C5Me4), 50.8 (br s, NMe2), 67.1 (CH2), 115.3 (C5Me4H CH), 119.3 (C6H4 CH), 121.8, 124.1, 124.8 (3 × C5Me4 Cipso), 125.6, 128.3 (2 × C6H4 CH), 155.0 (C6H4 C–N) ppm; signals due to C6H4 C–CH2, C5Me4 Cipso (both around 122.6 ppm) and C6H4 CH (around 129.7 ppm) obscured by the solvent signals. IR (KBr, cm−1): 2909 (m), 1591 (m), 1561 (w), 1471 (s), 1446 (m), 1370 (w) 1263 (s), 1232 (vs), 983 (w), 894 (m), 798 (w), 755 (m), 587 (s), 539 (w), 444 (w). M.p. = 240 °C. Anal. calc. for C36H50Cl2Zr2N4: C, 54.58; H, 6.35; N, 7.07. Found: C, 54.49; H, 6.29; N, 7.01.
[(η5-C5Me5){µ-NC6H4-2-(CH2NMe2)}ZrCl]2 (16). Orange crystals; yield 0.21 g (51%).
Tentatively assigned as 2 isomers (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
Major isomer, 1H NMR (300 MHz, toluene-d8) δ: 1.75 (s, 15H, C5Me5), 2.40 (s, 6H, NMe2), 3.47, 3.58 (2 × d, 2JHH = 15.3 Hz, 1H, CH2), 6.83–7.62 (m, C6H4 overlapping with the other isomer) ppm. 13C{1H} NMR (toluene-d8) δ: 11.2 (C5Me5), 47.3 (NMe2), 63.0 (CH2), 121.6, 122.7 (2 × C6H4 CH), 123.3 (C5Me5), 127.5, 130.3 (2 × C6H4 CH), 130.5 (C6H4 C–CH2), 150.2 (C6H4 C–N) ppm. Minor isomer, 1H NMR (300 MHz, toluene-d8) δ: 1.78 (s, 15H, C5Me5), 2.45 (s, 6H, NMe2), 3.66, 3.77 (2 × d, 2J = 15.5 Hz, 1H, CH2), 6.83–7.62 (m, C6H4 overlapping with the other isomer) ppm. 13C{1H} NMR (toluene-d8) δ: 11.6 (C5Me5), 47.5 (NMe2), 63.2 (CH2), 120.2, 123.4 (2 × C6H4 CH), 125.8 (C5Me5), 126.3, 131.2 (2 × C6H4 CH), 132.5 (C6H4 C–CH2), 148.1 (C6H4 C–N) ppm. IR (KBr, cm−1): 3054 (w), 2975 (m), 2941 (m), 2907 (s), 2858 (m), 2816 (m), 2771 (m), 1588 (m) 1563 (m), 1471 (s), 1441 (s), 1379 (m), 1363 (m), 1267 (s), 1230 (vs), 1172 (w), 1148 (w), 1097 (w), 1032 (m), 986 (w), 936 (w), 894 (m), 854 (m), 798 (w), 761 (vs), 735 (w), 721 (w), 635 (m), 578 (s), 546 (w), 507 (w), 484 (m), 433 (m). M.p. = 280 °C. Anal. calc. for C38H54Zr2Cl2N4: C, 55.64; H, 6.64; N, 6.83. Found: C, 55.58; H, 6.59; N, 6.78.
1H NMR (300 MHz, toluene-d8) δ: 1.22 (s, 3H, C5Me4 prox.), 2.09 (s, 6H, NMe2 of LNN), 2.17 (s, 3H, C5Me4 dist.), 2.26 (s, 3H, C5Me4 prox.), 2.27 (s, 3H, C5Me4 dist.), 2.37, 2.39 (2 × br s, 3H, NMe2 of LCN), 3.01 (d, 2JHH = 13.7 Hz, 1H, CH2 of LCN), 3.30, 3.45 (2 × d, 2JHH = 12.3 Hz, 1H, CH2 of LNN), 3.77 (d, 2JHH = 13.7 Hz, 1H, CH2 of LCN), 5.03 (s, 1H, C5Me4H), 6.54–6.61 (m, 1H, C6H4 of LNN), 6.84–7.16 (m, 6H, C6H4 of LCN and LNN), 7.67–7.72 (m, 1H, C6H4 of LCN), 8.71 (NH) ppm. 13C{1H} NMR (toluene-d8) δ: 11.2, 11.7 (2 × C5Me4 dist.), 13.0, 14.8 (2 × C5Me4 prox.), 44.8 (NMe2 of LNN), 47.6, 49.2 (NMe2 of LCN), 64.7 (CH2 of LNN), 70.3 (CH2 of LCN), 111.8 (C5Me4H CH), 117.8, 119.2 (2 × C6H4 CH of LNN), 120.1, 121.5, 121.7 (3 × C5Me4 Cipso), 123.0 (C6H4 C–CH2 of LNN), 123.6 (C5Me4 Cipso), 123.8, 127.1, 127.3 (3 × C6H4 CH of LCN), 128.0, 130.8 (2 × C6H4 CH of LNN), 143.2 (C6H4 CH of LCN), 146.9 (C6H4 C–CH2 of LCN), 153.3 (C6H4 C–N of LNN), 196.5 (C6H4 C–Hf of LCN) ppm. IR (KBr, cm−1): 3276 (w), 3043 (w), 2914 (m), 2855 (m), 2816 (m) 2773 (m), 1616 (m) 1600 (m), 1572 (w), 1486 (s), 1458 (s), 1363 (m), 1272 (s), 1254 (m), 1177 (w), 1148 (w), 1099 (m), 1043 (m), 1018 (s), 989 (w), 883 (m), 839 (m), 790 (m), 748 (vs), 724 (m), 645 (m), 614 (m), 507 (w), 430 (w). M.p. = 260 °C. Anal. calc. for C27H38ClHfN3: C, 52.43; H, 6.19; N, 6.79. Found: C, 52.38; H, 6.09; N, 6.72.
1H NMR (300 MHz, toluene-d8) δ: 1.91 (s, 15H, C5Me5), 2.06 (s, 6H, NMe2 of LNN), 2.36, 2.37 (2 × br s, 3H, NMe2 of LCN), 2.96 (d, 2JHH = 14.3 Hz, 1H, CH2 of LCN), 3.26, 3.45 (2 × d, 2JHH = 12.2 Hz, 1H, CH2 of LNN), 3.86 (d, 2JHH = 14.3 Hz, 1H, CH2 of LCN), 6.49–6.57 (m, 1H, C6H4 CH of LNN), 6.78–7.25 (m, 6H, C6H4 of LCN and LNN), 7.73–7.78 (m, 1H, C6H4 of LCN), 8.46 (NH) ppm. 13C{1H} NMR (toluene-d8) δ: 12.0 (C5Me5), 45.0 (NMe2 of LNN), 47.9, 48.1 (NMe2 of LCN), 64.5 (CH2 of LNN), 70.1 (CH2 of LCN), 117.7, 119.9 (C6H4 CH of LNN), 120.9 (C5Me5), 123.4 (C6H4 C–CH2 of LNN), 123.7, 126.6, 126.7, 129.2, 130.8 (C6H4 CH), 142.9 (C6H4 CH of LCN), 146.0 (C6H4 C–CH2 of LCN), 153.2 (C6H4 C–N of LNN), 196.7 (C6H4 C–Hf of LCN) ppm. IR (KBr, cm−1): 3235 (w), 2912 (vs), 2692 (w), 1636 (w), 1497 (m), 1457 (s), 1381 (m), 1244 (s), 983 (w), 930 (w), 751 (vs), 545 (m), 457 (s). Anal. calc. for C28H40ClHfN3: C, 53.16; H, 6.37; N, 6.64. Found: C, 53.08; H, 6.32; N, 6.55.
Conclusions
Two series of mononuclear complexes with terminal group 4 metal–amide bond(s), and dinuclear complexes with bridging fashion of an imide have been prepared and structurally characterized. All the described reactions were carried out in an equimolar ratio to maintain integrity of the studied reactions. Two different kinds of nitrogen ligands and two Cp ligands with different electronic and steric effects were employed in order to demonstrate variability of the group 4 metal coordination spheres. The solid-state structural study proved to be an essential tool for understanding of all ongoing processes. In summary, the amido complexes are monomeric, imido ones with LN ligand dinuclear. Similar arrangement of the substituents is adopted in cases of hafnium complexes with potentially N,N′-chelating amido ligand (LNN) and also the combination of zirconium complexes with LNN and bulkier Cp* ligands. The only exception is the dinuclear zirconium complex with LNN and less bulky Cp′ ligand, where the rare mutual arrangement of the same type of substituents is syn with respect to the central M2N2 plane. The results confirm anticipated differences in the chemical behavior and properties of group 4 metals showing an increasing stability of amido- and imido-complexes from titanium to hafnium.Acknowledgements
This research was supported by the Czech Science Foundation (Project No. P106/10/0924).References
- (a) M. H. Chisholm and I. P. Rothwell, in Comprehensive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon, Oxford, 1987, vol. 2, p. 161 and references therein Search PubMed; (b) M. F. Lappert, A. V. Protchenko, P. P. Power and A. L. Seeber, Metal Amide Chemistry, John Wiley & Sons, Hoboken, 2008 Search PubMed.
- P. D. Bolton and P. Mountford, Adv. Synth. Catal., 2005, 347, 355–366 CrossRef CAS.
- W. A. Nugent and B. L. Haymore, Coord. Chem. Rev., 1980, 31, 123–175 CrossRef CAS.
- L. H. Gade and P. Mountford, Coord. Chem. Rev., 2001, 216, 65–97 CrossRef.
- D. J. Mindiola, Acc. Chem. Res., 2006, 39, 813–821 CrossRef CAS PubMed.
- A. R. Fout, U. J. Kilgore and D. J. Mindiola, Chem.–Eur. J., 2007, 13, 9428–9440 CrossRef CAS PubMed.
- Y. H. Li, Y. H. Shi and A. L. Odom, J. Am. Chem. Soc., 2004, 126, 1794–1803 CrossRef CAS PubMed.
- I. Castillo and T. D. Tilley, J. Organomet. Chem., 2002, 643, 431–440 CrossRef.
- C. Lorber, R. Choukroun and L. Vendier, Eur. J. Inorg. Chem., 2006, 4503–4518 CrossRef CAS.
- (a) C. Lorber and L. Vendier, Inorg. Chem., 2011, 50, 9927–9929 CrossRef CAS PubMed; (b) C. Lorber and L. Vendier, Organometallics, 2010, 29, 1127–1136 CrossRef CAS.
- X. G. Yu, S. J. Chen, X. P. Wang, X. T. Chen and Z. L. Xue, Organometallics, 2009, 28, 4269–4275 CrossRef CAS.
- M. Said, D. L. Hughes and M. Bochmann, Dalton Trans., 2004, 359–360 RSC.
- J. R. Ascenso, C. G. de Azevedo, A. R. Dias, M. T. Duarte, I. Eleuterio, M. J. Ferreira, P. T. Gomes and A. M. Martins, J. Organomet. Chem., 2001, 632, 17–26 CrossRef CAS.
- (a) Y. I. Bai, M. Noltemeyer and H. W. Roesky, Z. Naturforsch., B: Chem. Sci., 1991, 46, 1357–1363 CrossRef CAS; (b) Y. Bai, H. W. Roesky and M. Noltemeyer, Z. Anorg. Allg. Chem., 1991, 595, 21–26 CrossRef CAS.
- (a) K. F. Liu, Q. L. Wu, W. Gao and Y. Mu, Dalton Trans., 2011, 4715–4721 RSC; (b) K. F. Liu, Q. L. Wu, W. Gao, Y. Mu and L. Ye, Eur. J. Inorg. Chem., 2011, 1901–1909 CrossRef.
- E. S. Cho, Y. C. Won, S. Y. Lee, B. Y. Lee, D. M. Shin and Y. K. Chung, Inorg. Chim. Acta, 2004, 357, 2301–2308 CrossRef CAS.
- B. M. Chapin, L. D. Hughs, J. A. Golen, A. L. Rheingold and A. R. Johnson, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2010, 66, M191–M193 CAS.
- Y. N. Bai, H. W. Roesky, M. Noltemeyer and M. Witt, Chem. Ber., 1992, 125, 825–831 CrossRef CAS.
- (a) K. H. Thiele, G. A. Hadi, J. Wunderle and H. Langhof, Z. Kristallogr., 1994, 209, 373–374 CAS; (b) P. Doufou, K. A. Abboud and J. M. Boncella, J. Organomet. Chem., 2000, 603, 213–219 CrossRef CAS; (c) A. C. Benjamin, A. S. P. Frey, M. G. Gardiner, C. L. Raston, B. W. Skelton and A. H. White, J. Organomet. Chem., 2008, 693, 776–780 CrossRef CAS; (d) V. Tabernero, T. Cuenca and E. Herdtweck, Eur. J. Inorg. Chem., 2004, 3154–3162 CrossRef CAS; (e) J. E. Bol, B. Hessen, J. H. Teuben, W. J. J. Smeets and A. L. Spek, Organometallics, 1992, 11, 1981–1983 CrossRef CAS; (f) K. E. Duplooy, U. Moll, S. Wocadlo, W. Massa and J. Okuda, Organometallics, 1995, 14, 3129–3131 CrossRef CAS.
- (a) W. J. Grigsby, M. M. Olmstead and P. P. Power, J. Organomet. Chem., 1996, 513, 173–180 CrossRef CAS; (b) S. J. Lancaster, S. Al-Benna, M. Thornton-Pett and M. Bochmann, Organometallics, 2000, 19, 1599–1608 CrossRef CAS; (c) K. Weitershaus, H. Wadepohl and L. H. Gade, Organometallics, 2009, 28, 3381–3389 CrossRef CAS; (d) A. Abarca, P. Gomez-Sal, A. Martin, M. Mena, J. M. Poblet and C. Yelamos, Inorg. Chem., 2000, 39, 642–651 CrossRef CAS PubMed; (e) K. Kaleta, P. Arndt, T. Beweries, A. Spannenberg, O. Theilmann and U. Rosenthal, Organometallics, 2010, 29, 2604–2609 CrossRef CAS; (f) H. Tsurugi, H. Nagae and K. Mashima, Chem. Commun., 2011, 47, 5620–5622 RSC; (g) A. K. Hughes, S. M. B. Marsh, J. A. K. Howard and P. S. Ford, J. Organomet. Chem., 1997, 528, 195–198 CrossRef CAS; (h) P. J. Walsh, F. J. Hollander and R. G. Bergman, J. Am. Chem. Soc., 1988, 110, 8729–8731 CrossRef CAS; (i) A. M. Baranger, P. J. Walsh and R. G. Bergman, J. Am. Chem. Soc., 1993, 115, 2753–2763 CrossRef CAS; (j) Z. Li, J. Huang, T. Yao, Y. Qian and M. Leng, J. Organomet. Chem., 2000, 598, 339–347 CrossRef CAS; (k) J. F. Eichler, O. Just and W. S. Rees, Phosphorus, Sulfur Silicon Relat. Elem., 2004, 179, 715–726 CrossRef CAS; (l) G. C. Bai, H. W. Roesky, H. J. Hao, M. Noltemeyer and H. G. Schmidt, Inorg. Chem., 2001, 40, 2424–2426 CrossRef CAS PubMed; (m) Y. Bai, H. W. Roesky, H. G. Schmidt and M. Noltemeyer, Z. Naturforsch., B: Chem. Sci., 1992, 47, 603–608 CrossRef CAS; (n) J. D. Selby, M. Feliz, A. D. Schwarz, E. Clot and P. Mountford, Organometallics, 2011, 30, 2295–2307 CrossRef CAS.
- (a) J. A. Pool, E. Lobkovsky and P. J. Chirik, Nature, 2004, 427, 527–530 CrossRef CAS PubMed; (b) J. N. Armor, Inorg. Chem., 1978, 17, 203–213 CrossRef CAS; (c) J. A. Pool, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2003, 125, 2241–2251 CrossRef CAS PubMed; (d) W. H. Bernskoetter, J. A. Pool, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2005, 127, 7901–7911 CrossRef CAS PubMed; (e) D. J. Knobloch, E. Lobkovsky and P. J. Chirik, Nat. Chem., 2010, 2, 30–35 CrossRef CAS PubMed; (f) W. H. Bernskoetter, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2005, 127, 14051–14061 CrossRef CAS PubMed; (g) W. H. Bernskoetter, A. V. Olmos, E. Lobkovsky and P. J. Chirik, Organometallics, 2006, 25, 1021–1027 CrossRef CAS; (h) T. E. Hanna, W. H. Bernskoetter, M. W. Bouwkamp, E. Lobkovsky and P. J. Chirik, Organometallics, 2007, 26, 2431–2438 CrossRef CAS; (i) M. Hirotsu, P. P. Fontaine, P. Y. Zavalij and L. R. Sita, J. Am. Chem. Soc., 2007, 129, 12690–12692 CrossRef CAS PubMed; (j) S. P. Semproni, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2011, 133, 10406–10409 CrossRef CAS PubMed.
- (a) T. W. Graham, J. Kickham, S. Courtenay, P. R. Wei and D. W. Stephan, Organometallics, 2004, 23, 3309–3318 CrossRef CAS; (b) A. Decker, D. Fenske and K. Maczek, Angew. Chem., Int. Ed., 1996, 35, 2863–2866 CrossRef CAS; (c) H. W. Roesky, Y. Bai and M. Noltemeyer, Angew. Chem., Int. Ed., 1989, 28, 754–755 CrossRef.
- A. Havlík, M. Lamač, J. Pinkas, Z. Padělková, A. Růžička, R. Gyepes and M. Horáček, J. Organomet. Chem., 2012, 719, 64–73 CrossRef.
- A. Havlík, M. Lamač, J. Pinkas, V. Varga, A. Růžička, R. Olejník and M. Horáček, J. Organomet. Chem., 2015, 786, 71–80 CrossRef.
- H. J. Reich, WINDNMR Program, 2002 Search PubMed.
- (a) H. Lang, R. Packheiser and B. Walfort, Organometallics, 2006, 25, 1836–1850 CrossRef CAS; (b) R. Packheiser, P. Ecorchard, T. Ruffer and H. Lang, Organometallics, 2008, 27, 3534–3546 CrossRef CAS; (c) R. Packheiser, A. Jakob, P. Ecorchard, B. Walfort and H. Lang, Organometallics, 2008, 27, 1214–1226 CrossRef CAS.
- (a) P. Pyykko and M. Atsumi, Chem.–Eur. J., 2009, 15, 186–197 CrossRef PubMed; (b) P. Pyykko and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef PubMed; (c) P. Pyykko, S. Riedel and M. Patzschke, Chem.–Eur. J., 2005, 11, 3511–3520 CrossRef PubMed.
- (a) J. Bareš, P. Richard, P. Meunier, N. Pirio, Z. Padělková, Z. Černošek, I. Císařová and A. Růžička, Organometallics, 2009, 28, 3105–3108 CrossRef; (b) Z. Padělková, A. Havlík, P. Švec, M. S. Nechaev and A. Růžička, J. Organomet. Chem., 2010, 695, 2651–2657 CrossRef.
- (a) J. Bareš, V. Sourek, Z. Padělková, P. Meunier, N. Pirio, I. Císařová, A. Růžička and J. Holeček, Collect. Czech. Chem. Commun., 2010, 75, 121–131 CrossRef; (b) H. Vaňkátová, L. Broeckaert, F. De Proft, R. Olejník, J. Turek, Z. Padělková and A. Růžička, Inorg. Chem., 2011, 50, 9454–9464 CrossRef PubMed.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CAS.
- P. Coppens, Crystallographic Computing, Munksgaard, Copenhagen, 1970 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343–350 CrossRef.
- G. M. Sheldrick and T. R. Schneider, Methods Enzymol., 1997, 277, 319–343 CAS.
- P. Vandersluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Tables of selected crystallographic parameters for 3, 4, 5, 6, 7, 8, 8′, 10, 11, 12, 13, 14, 15, 16, 17. Figures of molecular structure and selected interatomic distances and angles for 3, 4, 14. Eyring plots for 8 and 9. An expanded region of 1H NMR spectra of 8 in the temperature range from −35 °C to 55 °C. CCDC 1044550–1044564. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra07742g |
| This journal is © The Royal Society of Chemistry 2015 |