
New photocatalyst for allylic aliphatic C–H bond activation and degradation of organic pollutants: Schiff base Ti(iv) complexes†
Abstract
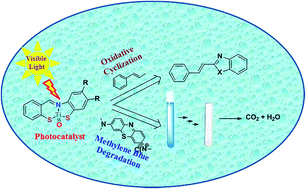
Schiff base metal complexes have attracted significant attention due to their alternative applications in the environment, such as the mineralization of organic pollutants to less harmful byproducts and oxygen generation via such processes as photosynthesis. The Ti4+ complexes obtained from Schiff base ligands reacted with donor atoms such as S and N under solvothermal conditions. These complexes were characterized using microanalysis, conductivity studies, and different spectral techniques. These data reveal that the compounds show distorted octahedral geometries with ligand coordination via azomethine nitrogen and thiol sulfur atoms. Oxidation of the allylic methyl group examined the photocatalytic activity of [Ti(L)O] under ambient conditions and oxidative cyclization under visible light irradiation. The Ti(DCMPPT)O complex is a very efficient catalyst due to the very short span of time it takes to produce aldehydes from allylic compounds. Aldehydes readily react with 2-aminophenol or 2-aminobenzenethiol to produce (E)-2-styrylbenzo[d]oxazoles and (E)-2-styrylbenzo[d]thiazoles due to the suitability of the bandgap energy to the generation of ˙OH radicals during the catalytic reaction. This results in a higher oxidation rate. [Ti(DCMPPT)O] is a very efficient photocatalyst for the degradation of MB, due to its large surface area which suggests a lower recombination energy mechanism.
 Please wait while we load your content...
Please wait while we load your content...

