
Ordered mesoporous crystalline aluminas from self-assembly of ABC triblock terpolymer–butanol–alumina sols†
 a
a
Abstract
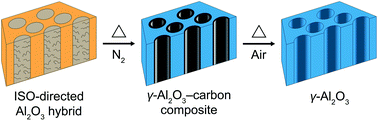
A one-pot synthesis approach is described to generate ordered mesoporous crystalline γ-alumina–carbon composites and ordered mesoporous crystalline γ-alumina materials via the combination of soft and hard-templating chemistries using block copolymers as soft structure-directing agents. Periodically ordered alumina hybrid mesostructures were generated by self-assembly of a poly(isoprene)-block-poly(styrene)-block-poly(ethylene oxide) terpolymer, n-butanol and aluminum tri-sec-butoxide derived sols in organic solvents. The triblock terpolymer was converted into a rigid carbon framework during thermal annealing under nitrogen to support and preserve the ordered mesoporous crystalline γ-alumina–carbon composite structures up to 1200 °C. The carbon matrix was subsequently removed in a second heat treatment in air to obtain ordered mesoporous crystalline γ-alumina structures. Such thermally stable, highly crystalline, and periodically ordered mesoporous ceramic and ceramic–carbon composite materials may be promising candidates for various high temperature catalysis, separation, and energy-related applications.
 Please wait while we load your content...
Please wait while we load your content...

