
Extending the limits of emulsifier-free emulsion polymerization to achieve small uniform particles
Abstract
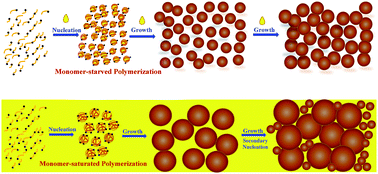
Conventional emulsifier-free emulsion polymerization often produces large particles, which are hardly uniform in the case of partially water-soluble monomers. In this research, preparation of uniform polymeric particles was attempted using monomer-starved semicontinuous emulsifier-free emulsion polymerization. Two monomers, with different water solubility, vinyl acetate (VA) and butyl acrylate (BA), were used as model monomers. The continuous fall in the number of particles (Np) usually observed in the early stage of ab initio emulsifier-free emulsion polymerization transformed to a rapidly rising Np for the semicontinuous operation. The semicontinuous process was found to be capable of producing uniform particles that are much smaller than those achievable via conventional process. This was attributed to the enhanced particle stability achieved by reducing the rate of particle growth and by prohibiting secondary nucleation via the aqueous phase being starved with the monomer. The formation of large number of small particles under monomer-starved conditions was found to be a feasible strategy to prevent secondary nucleation. This allows to apply this one-stage methodology to synthesize a wide range of uniform polymeric particles. The particle sizes decreased with decreasing monomer feed rate, while their uniformity was maintained. However, there is a minimum rate of monomer addition, depending on monomer solubility in water and polymerization conditions, below which uniform particles may not be achieved.
 Please wait while we load your content...
Please wait while we load your content...

