
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation on the electrochemistry and cytotoxicity of the natural product marcanine A and its synthetic derivatives†
Nadine
Jacobs
a,
Steffen
Lang
b,
Robin
Panisch
c,
Gunther
Wittstock
a,
Ulrich
Groth
b and
Hamid R.
Nasiri
*c
aCarl von Ossietzky University of Oldenburg, Faculty of Mathematics and Natural Sciences, Institute of Chemistry, D-26111 Oldenburg, Germany
bFachbereich Chemie und Konstanz Research School Chemical Biology, Universität Konstanz, Postfach M-720, D-78457 Konstanz, Germany
cJohann Wolfgang Goethe-University Frankfurt, Max-von-Laue-Straße 7, D-60438 Frankfurt am Main, Germany. E-mail: Nasiri@nmr.uni-frankfurt.de
First published on 29th June 2015
Abstract
The electrochemistry and cytotoxicity of marcanine A were investigated by electrochemical, computational and cellular studies. To enable a structure–toxicity-relationship of the natural product, eleven novel synthetic derivatives with different electrochemical properties were synthesized and tested. Derivative 5 revealed a GI50 in the low μM range, being more active than the actual natural product. A clear correlation was found between the experimental and the calculated data.
Introduction
Marcanine A 1 was first isolated in 1999 from the stem bark of Goniothalamus marcanii from Thailand.1 This natural product possesses remarkable biological activity including antimalarial activity2 and more importantly, cytotoxicity against various tumor cell lines.3 Marcanine A 1, together with the related natural products kalasinamide 2 and geovanine 3 belong to the azaanthracenone natural product family (Fig. 1).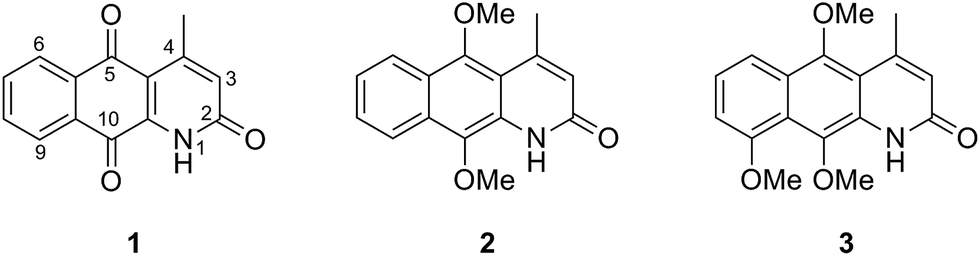 | ||
| Fig. 1 Constitution of the azaanthracenone natural products: marcanine A 1, kalasinamide 2 and geovanine 3. | ||
Recently, we and others reported the synthesis of these azaanthracenone natural products and investigated their cytotoxic effect against different cell lines.3,4 Interestingly, while marcanine A 1 has a strong cytotoxic effect (HeLa, a concentration required to inhibit cell growth by 50% (GI50): 0.75 ± 0.03 μM; Hep, GI50: 1.54 ± 0.78 μM), the other two natural products (2 and 3) were totally inactive.3
The underlying mechanism of the cytotoxic effect of quinones is complex and has been attributed to various events such as redox cycling, mitochondrial dysfunction,5,6 intercalation into DNA, inhibition of the DNA topoisomerase complex and alkylation via Michael addition. The cytotoxic properties of quinones are of great interest in current cancer therapy.7 Daunorubicin, mitomycin C and β-lapachone are examples of quinone-based drugs, which are currently in clinical use or in clinical trials for treatment of a variety of cancers in human.7,8 In particular, azaanthraquinones were tested for their ability to bind DNA and interfere with DNA polymerase activity in vitro.9 The mode of action of azaanthraquinones was determined to be intercalation into the DNA double strand and inhibition of DNA supercoiling.9
It has been postulated that the cytotoxicity is mediated through semiquinone radical species, which are formed during the quinone to quinol conversion and vice versa.10 Here, the cytotoxicity is highly dependent on the electrochemical potential of the initially formed radical species.11 For all mechanisms involved in cytotoxicity, the redox potential of the quinone under investigation is believed to play a significant role.
We postulate that the cytotoxic properties could be related to the presence of a quinone scaffold, which exists in marcanine A 1 but not in the other, structurally similar natural products (2-3). In order to investigate the role of the quinone moiety as a relevant scaffold in azaanthracenone natural products, manipulation of the redox potential of the quinone motif was performed to explore the structure–toxicity-relationship with the ultimate aim of designing novel molecules with improved anticancer activity. Ten marcanine A derivatives (4–14) with different substitution patterns were synthesized with variation in size and electrochemical properties of the functional groups attached to the azaanthracenone scaffold (Fig. 2). The electrochemistry of these derivatives was investigated by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) and compared to calculations using density functional theory (DFT). A clear correlation was found between experimentally determined redox potentials and theoretically calculated electron affinities of the synthesized derivatives. These derivatives were further tested against two different cancer cell lines. The growth inhibition data are discussed in light of measured and calculated electrochemical data.
 | ||
| Fig. 2 Constitution of the synthesized marcanine A derivatives (4–14) used in this study. | ||
Results and discussion
Chemistry
In order to investigate the influence of substituents on the cytotoxicity and electrochemistry of marcanine A 1, the methyl-group at the 4-position was removed (4), or systematically altered with different substituents (5–9) (Fig. 2). In the first step, we changed the electrochemical properties of marcanine A 1 by introducing electron donating (5, 9) and electron withdrawing groups (8). In the second variation step, we changed the bulkiness of the structure by introducing phenyl-7 and anisyl-6 substituents. Compound 9 was synthesized as a positional isomer of 5, with the methoxy-group at the 5-position, in order to investigate the influence of the position of the substituents.In order to estimate the contribution of the lactam scaffold to cytotoxicity and electrochemistry of marcanine A 1, analogues of 6 and 7 were prepared with a trapped lactam moiety i.e.10 and 11. Finally, the lactam moiety of marcanine A 1 was further investigated by replacing the keto group at the 2-position by hydrogen 12, methyl 13 and chlorine 14 substituents. An overview of the multi-step synthesis of these compounds is included in ESI† and described in more detail elsewhere.12
Electrochemistry
To explore the electrochemistry of marcanine A 1 and its synthesized derivatives (4–14) cyclic voltammetry (CV) and differential pulse voltammetry (DVP) experiments were performed using a conventional three-electrode cell in deaerated solutions. All experiments were conducted in the aprotic solvent dimethylformamide (DMF) with tetrabutylammonium hexafluorophosphate (TBAPF6) as supporting electrolyte on platinum working and counter electrodes. DMF was used as an aprotic solvent to mimic the nonpolar environment inside the cells. A Haber Luggin dual reference electrode13 (HLDRE, ESI†) was used as a reference electrode. The formal potential (E°′) was calculated using E°′ = 0.5(Epa + Epc) for CV experiments, where Epa represents the anodic peak and Epc the corresponding cathodic peak potentials. All given potentials are referred to the ferrocene/ferrocenium redox couple. The reversibility and scan rate dependencies were evaluated by running each experiment at different scan rates and for several cycles (ESI†). DPV was used in addition to CV to independently determine the formal potential (E°′) of the tested compounds. As an example the CV and DPV of azaanthracenone 5, the most potent derivative, are shown in Fig. 3. The CV displays a quasi-reversible one-electron reduction (E°′1 = −0.99 V) indicated by a symmetric shape of the curve. In contrast, the second reduction process (E°′2 = −1.73 V) is irreversible due to the lack of an oxidation peak. The first reduction step represents the formation of a semi-quinone radical, the second reduction step represents the forming of the quinone dianion species.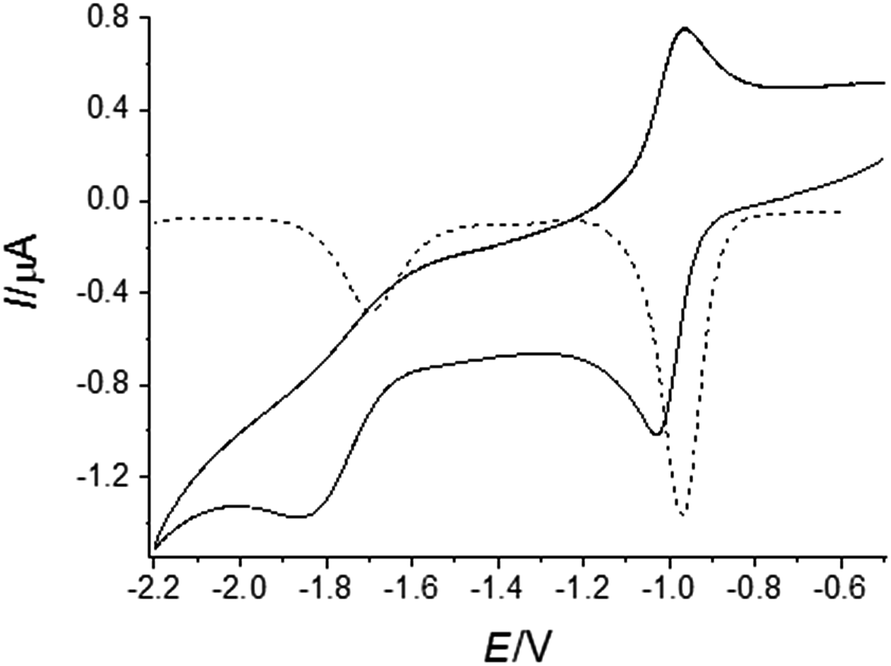 | ||
| Fig. 3 CV (__) and DPV (--) of azaanthracenone (5) at 1 mM concentration, dissolved in DMF with tetrabutylammonium hexafluorophosphate (TBAPF6) as supporting electrolyte on platinum working and counter electrodes at a scan rate of 0.1 V s−1 and DPV with an amplitude of 0.05 V. The first cathodic peak (Epc = −1.03 V) originates from the reduction of the quinone to the semi-quinone radical and the anodic peak (Epa = −0.96 V) corresponds to the reoxidation of the semiquinone species back to the quinone form. | ||
All investigated derivatives of marcanine A 1 show similar qualitative behavior (ESI†). The formal potentials (E°′) and calculated electron affinities (Ea) of tested compounds are listed in Table 1.
| E°′1 [V] | E°′2 [V] | E a [eV] | |
|---|---|---|---|
| a Data according to Díaz-Guerra et al.14 | |||
| 1 | −1.016 | −1.730 | −2.302 |
| 4 | −0.95a | −1.58a | −2.344![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| 5 | −0.997 | −1.735 | −2.229 |
| 6 | −0.999 | −1.741 | −2.312 |
| 7 | −1.008 | −1.746 | −2.352 |
| 8 | −0.919 | −1.527 | −2.490 |
| 9 | −1.125 | −1.695 | −2.190 |
| 10 | −1.255 | −1.885 | −1.794 |
| 11 | −1.255 | −2.016 | −1.832 |
| 12 | −1.319 | −1.883 | −1.799 |
| 13 | −1.328 | −1.937 | −1.731 |
| 14 | −1.145 | −1.640 | −2.048 |
As expected, compound 8 with a chlorine electron withdrawing group directly attached to the quinone core revealed the highest formal potential (E°′1 = −0.91 V) and was therefore easier to reduce. By contrast, compounds (10-13), exhibit the lowest midpoint potential, indicating that removing or trapping the lactam motif by methylation decreased the electron affinity of the quinone scaffold. Experimentally measured formal potentials for compounds 5–7 appeared in the same range. Interestingly, compounds 5 and 9, both with an electron donating methoxy substituent, were separated experimentally by 0.128 V. This result signifies that the methoxy substituent at the C-3 position stabilizes the radical anion in 9 better compared to 5 and reduces the electron affinity of the quinone moiety.
DFT calculations
The first electron reduction process is the more relevant parameter, which has been extensively used in the literature for the correlation of electrochemistry and biological activity.15 For this reason, only adiabatic electron affinities (Ea) corresponding to the first reduction step were calculated for the series of synthesized azaanthraquinones (4–14) using DFT (Table 1). We have recently demonstrated that this particular method can be applied as an appropriate tool for the computational investigation of the electrochemical properties of 1,4-naphthoquinones.16As shown in Fig. 4, an excellent correlation is observed between calculated electron affinities (Ea) and experimental formal potentials E°′1.
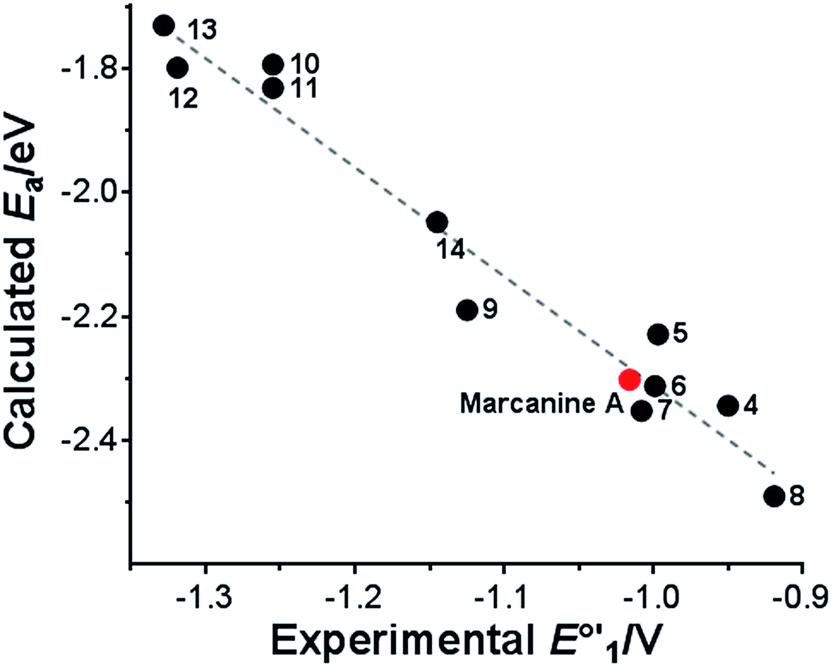 | ||
| Fig. 4 Correlation of experimental formal potentials E°′1 with calculated adiabatic electron affinities Ea. Ea = −1.75 × E°′1−4.06; R2 = 0.96. | ||
Not only were the compounds that were investigated experimentally by electrochemical methods included in the computational studies, we were also able to accurately calculate the electron affinities of a number of naturally occurring quinones and quinoid compounds with different substitution patterns. Therefore, our computational approach does not only allow for the rationalization of experimental findings, but can also be applied to predict the properties of novel quinones (ESI†).
In agreement with our previous findings,16 the calculated electron affinities are clearly related to both the expected electronic effects and the position of the substituents. While quinones with electron donating substituents such as R = NH2, OMe have comparatively low electron affinities, strongly electron withdrawing substituents like R = F, Cl, CF3, give rise to higher values. Quinones with substituents at the 3-position have lower Ea than 4-substituted quinones. However, this trend is reversed if R is a bulky group, e.g. R = C6H4OMe, CF3, Ph, probably as a result of the unfavourable proximity to the adjacent carbonyl group. The presence of a hydroxyl group next to a carbonyl moiety increases Ea as demonstrated by the comparison of marcanine D with dielsiquinone and lambertellin with 9H-lambertellin (ESI†). Methylation of the amide group decreases the electron affinity (marcanine B Ea = −2.099 eV, dielsiquinone Ea = −2.229 eV) while the exchange of the lactam group with a lactone functionality results in higher values (lambertellin Ea = −2.545 eV, lactone derivative Ea = −2.437 eV).
Biological testing
The antiproliferative activity of the newly synthesized compounds against human HeLa melanoma and Hep G2 hepatocellular carcinoma cell lines was tested using the alamarBlue® assay (Life Technologies). The growth inhibition (GI50) values were determined by a nonlinear regression. cis-Platin and doxorubicin were used as positive controls. The ability of marcanine A 1 and its derivatives (4–14) to inhibit the growth of both cancer cell lines is represented in Fig. 5. Compounds 6, 10 and 11 were poorly soluble in aqueous media and were therefore excluded from further biological testing. | ||
| Fig. 5 Growth inhibition (GI50) values of marcanine A 1 and its derivatives (4–14) against HeLa and Hep cancer cell lines determined in an alamarBlue® assay. Results are the average of three measurements. | ||
Compound 5 was the most active compound in our assay with GI50 values in the low micromolar range (Fig. 5). This compound showed an improved cytotoxic activity compared to the actual natural product marcanine A 1 (HeLa GI50: 0.75 ± 0.03 μM; Hep GI50: 1.54 ± 0.78 μM). Interestingly, its positional isomeric compound 9, with the methoxy-substituent at C3 was ten-fold (against HeLa) and fivefold (against Hep) less active compared to 5. Compounds 5 and 9 bearing an electron donating methoxy group at C4 and C3 position, were more potent compared to the electron withdrawing chloro-substituent in compound 8. Hydrophobic and sterically hindered derivative 7 was less active compared to derivatives 5 and 9 with smaller and polar substituents. Compounds 5, 7, 8 and 9 with a lactam scaffold were more potent against both cell lines than the corresponding compounds 12, 13 and 14 lacking this structural motif.
There is also an interdependency of antiproliferative activity of tested compounds with their intrinsic electrochemical properties (Fig. 6). Compounds (12–14) with a pyridine scaffold and lower formal potentials E°′1 showed also the lowest cytotoxic effect. Following the trend, compounds 5–9 with the 2-pyridone scaffold and higher formal potentials E°′1 compared to the pyridine series were also several times more active in the cellular assay (Fig. 6). Similar correlation was observed between the antiproliferative properties in Hep cells, the measured and calculated electrochemical affinities (ESI†).
 | ||
| Fig. 6 Interdependency of antiproliferative activity of tested compounds with their intrinsic electrochemical properties. Correlation of calculated electron affinities and measured GI50 values in HeLa cells for pyridine and 2-pyridone series. | ||
Conclusions
By comparing the cytotoxicity of marcanine A 1 and other related natural products (2, 3), we identified the quinone scaffold as a potential pharmacophore. A focused set of marcanine A 1 derivatives (4–14) were designed, synthesized and tested. All of the designed compounds contained the quinone motif with different substituents to fine tune the redox behaviour and cytotoxicity of this class of molecules. The testing revealed a clear link between the presence of the redox active quinoid system and the observed cytotoxicity. In an attempt to predict the biological activities of quinones and quinoid compounds, the electron affinities were calculated by DFT methods. A clear correlation was observed between experimentally measured E°′ and theoretically calculated Ea values.The cellular results were rationalized by comparison with the electrochemical findings.
The presented approach provides strong support for the incorporation of electrochemical studies into the common structure–activity-relationship (SAR) studies of redox-active small molecules of biological or medical importance. Furthermore, this study supports a rational approach towards discovery, design and prediction of novel redox-based anticancer agents.
Acknowledgements
We thank Prof. Dr Harald Schwalbe and Dr Sina Berndl for helpful discussions and critical reading of this manuscript. We thank Dr Thomas Huhn for compound logistics and Mrs Malin Bein for conducting the tests on cytotoxicity. S.L. thanks the Studienstiftung des Deutschen Volkes for a doctoral fellowship and the Konstanz Research School Chemical Biology (KoRS-CB) for scientific support.References
- N. Soonthornchareonnon, K. Suwanborirux, R. Bavovada, C. Patarapanich and J. M. Cassady, J. Nat. Prod., 1999, 62, 1390–1394 CrossRef CAS.
- C. Ichino, N. Soonthornchareonnon, W. Chuakul, H. Kiyohara, A. Ishiyama, H. Sekiguchi, M. Namatame, K. Otoguro, S. Omura and H. Yamada, Phytother. Res., 2006, 20, 307–309 CrossRef CAS PubMed.
- S. Lang and U. Groth, Angew. Chem., Int. Ed., 2009, 48, 911–913 CrossRef CAS PubMed.
- M. N. Gandy and M. J. Piggott, J. Nat. Prod., 2008, 71, 866–868 CrossRef CAS PubMed.
- A. Emadi, A. Le, C. J. Harwood, K. W. Stagliano, F. Kamangar, A. E. Ross, C. R. Cooper, C. V. Dang, J. E. Karp and M. Vuica-Ross, Bioorg. Med. Chem., 2011, 19, 7057–7062 CrossRef CAS PubMed.
- A. Emadi, A. E. Ross, K. M. Cowan, Y. M. Fortenberry and M. Vuica-Ross, PLoS One, 2010, 5(5), e10846, DOI:10.1371/journal.pone.0010846.
- G. Escherich, M. Zimmermann and G. Janka-Schaub, Pediatr. Blood Cancer, 2013, 60, 254–257 Search PubMed.
- M. S. Bentle, E. A. Bey, Y. Dong, K. E. Reinicke and D. A. Boothman, J. Mol. Histol., 2006, 37, 203–218 CrossRef CAS PubMed.
- G. Burckhardt, A. Walter, H. Triebel, K. Storl, H. Simon, J. Storl, A. Opitz, E. Roemer and C. Zimmer, Biochemistry, 1998, 37, 4703–4711 CrossRef CAS PubMed.
- R. Pethig, P. R. Gascoyne, J. A. McLaughlin and A. Szent-Györgyi, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 129–132 CrossRef CAS.
- A. Brisach-Wittmeyer, A. S. Souna Sido, P. Guilini and L. Desaubry, Bioorg. Med. Chem. Lett., 2005, 15, 3609–3610 CrossRef CAS PubMed.
- S. Lang, Synthese und biologische Evaluierung von Azaanthracenonen, PhD Thesis, Cuvillier Verlag Göttingen, 2009, ISBN: 9783869551289.
- B. Gollas, B. Kraub, B. Speiser and H. Stahl, Curr. Sep., 1994, 13, 42–44 CAS.
- L. M. Díaz-Guerra, B. Ocaña, J. M. Pérez, C. Avendaño, M. Espada, J. C. Menéndez, M. T. Ramos, M. A. Ruiz, J. M. Pingarrón, D. Salvatierra and C. Jaime, Bull. Soc. Chim. Belg., 1995, 104, 683–690 CrossRef.
- E. A. Hillard, F. C. de Abreu, D. C. M. Ferreira, G. Jaouen, M. O. F. Goulart and C. Amatore, Chem. Commun., 2008, 2612–2628 RSC.
- H. R. Nasiri, R. Panisch, M. G. Madej, J. W. Bats, C. R. Lancaster and H. Schwalbe, Biochim. Biophys. Acta, 2009, 1787, 601–608 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra11078e |
| This journal is © The Royal Society of Chemistry 2015 |