
The role of structural and electronic factors in shaping the ambipolar properties of donor–acceptor polymers of thiophene and benzothiadiazole†
Abstract
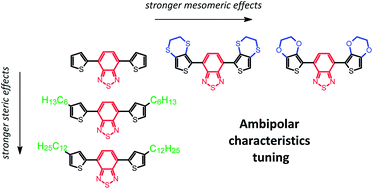
The influence of different thiophene donor units on electrochemical and spectroscopic properties of benzothiadiazole based donor–acceptor π-conjugated organic materials is studied. Two different structure modification vectors of the donor units are being considered – one addressing the intermolecular interactions through off-conjugation side chain architecture, and the other focusing on intramolecular interactions tuned by in-conjugation substituents. Electrochemical and simultaneous in situ EPR-UV-Vis-NIR spectroelectrochemical studies of the oxidative (p-) and reductive (n-) doping processes, which are responsible for the optoelectronic properties of these materials, revealed their disparate course and dissimilar effects of redox reactions of the conjugated π-bond. While p-doping prevalent species were found to comprise intensively interacting spin bearing and spinless charge carriers, the n-doping state was found to involve only one type of negatively charged carrier, with spin carrying species being selectively generated at due cathodic potentials. No spin pairing of these negative polarons was observed with their increasing population behaving like a collection of localised charge carriers. Qualitative and quantitative comparisons between the p- and n-doping carrier populations provided independent support for the spin pairing phenomena of positive charge carriers. Steric effects of varying alkyl side chain substitution have demonstrated predominant impact on the electrochemical properties of investigated polymers, and, thereto related, stability of n-doped state, while mesomeric effects of different 3,4-ethylenechalcogenide thiophene functionalities have been found to shape the energy level related spectral properties of these polymers, with particular reference to p-doping induced charged states. These findings provide new insights into the factors requiring attention during structure tailoring of donor–acceptor assemblies for organic optoelectronic applications.
 Please wait while we load your content...
Please wait while we load your content...

