
A facile route to prepare few-layer graphene/polyamide 6 nanocomposites by liquid reactive extrusion†
Xubing Fua,
Xingke Zhaob,
Dongguang Yanc,
Dajiang Zhaoa,
Jiao Lic and
Guisheng Yang*abd
aSchool of Chemistry and Chemical Engineering, Hefei University of Technology, Hefei, Anhui 230009, China. E-mail: ygs1211@sina.com; Fax: +86-21-64881869; Tel: +86-21-64900066
bDepartment of Polymer Science and Engineering, Zhejiang University, Hangzhou, Zhejiang 310027, China
cSchool of Material Science and Engineering, Jiangsu University of Science and Technology, Zhenjiang, Jiangsu 212003, China
dShanghai Genius Advanced Material Co., Ltd, Shanghai 201109, China
First published on 8th September 2015
Abstract
In this work, a liquid reactive extrusion process was developed to prepare few-layer graphene (FLG)/polyamide 6 (PA6) nanocomposites with low loading of nanofillers. Mass graphene flakes were fabricated by a re-expansion and exfoliation approach. The structure and morphology of FLG and FLG/PA6 nanocomposites were characterized by Raman spectroscopy, X-ray photoelectron spectroscopy, high-resolution transmission electron microscopy, field-emission scanning electron microscopy and Fourier-transformed infrared analysis. Moreover, the mechanical and crystallization properties of the PA6 and FLG/PA6 nanocomposites were also observed.
1. Introduction
There are several allotropes of carbon that have different spatial dimensions, including 0D (zero dimensional) fullerene, 1D nanotubes, 2D graphene, 3D graphite; as well as other crystalline and non-crystalline forms.1–3 In the case of graphene, a 2D crystalline single layer sheet of sp2-bonded carbon atoms are arranged in a honeycomb lattice, and has attracted great interest because of its unique and outstanding mechanical, thermal, electrical and chemical properties, such as carrier mobilities as high as ∼200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1, a thermal conductivity up to ∼5000 W m−1 K−1 and a Young's modulus of ∼1.1 TPa.4–11 Since graphene can improve the mechanical, electrical, thermal and other properties of polymer composites, graphene/polymer nanocomposites are very promising when graphene is used as nanofillers.12,13
000 cm2 V−1 s−1, a thermal conductivity up to ∼5000 W m−1 K−1 and a Young's modulus of ∼1.1 TPa.4–11 Since graphene can improve the mechanical, electrical, thermal and other properties of polymer composites, graphene/polymer nanocomposites are very promising when graphene is used as nanofillers.12,13
Three main methods are commonly used to prepare graphene/polymer nanocomposites: in situ polymerization, solvent blending and melt blending.10,12,14 During in situ polymerization, graphene is mixed with the liquid monomer, then an initiator is added and the polymerization reaction occurs by heat or radiation.15 For example, O'Neill et al. (2014) prepared graphene/PA6 nanocomposites via in situ ring opening polymerization of ε-caprolactam in the presence of single layer graphene oxide (GO), and found that this method can yield a good dispersion of GO sheets in the PA6 matrix.16 Ding et al. (2014) synthesized reduced graphene oxide (RGO)/PA6 nanocomposite also by in situ polymerization and found that the thermal conductivity improved.17 Compared to the in situ polymerization, solvent blending is a simpler method. In this method, graphene is first dispersed in a suitable solvent, and the polymer is incorporated secondly, the solvent is later removed by evaporation or distillation. Tkalya et al. (2010) prepared polystyrene/graphene nanocomposites by solvent blending and studied their conductivity. It was found that this method achieved a fine dispersion of graphene in the polymer matrices.18 Unlike the above two methods, melt blending is the most practical approach. This method involves the direct addition of graphene into the molten polymer using a screw extruder or injection mould. Oana et al. (2014) fabricated polymer composites reinforced with exfoliated graphene layers solely via melt blending, and found enhancement in the tensile strength of more than 40% compared to the pristine polymer matrix.19 Among these three methods, in situ polymerization and solvent blending usually lead to a good dispersion of graphene sheets in the polymer matrices while melt blending causes poor dispersion.20 Because of this, most graphene/polymer nanocomposites are fabricated by in situ polymerization and solvent blending. From an industrial point of view, melt blending is more suitable for large-scale production as it does not require large amounts of organic solvents and the preparation is simple. However, melt blending results in many cases in an incomplete dispersion of graphene in the polymer matrix.21,22 Thus, a new approach needs to be found, which may not only lead a good dispersion of graphene in the polymer, but will be suitable for industrial production.
A possible solution for this could be in situ polymerization that can also be performed in an extruder. Nanofillers and other components, such as activators and initiators, can be mixed into the liquid monomer, and the nanofillers can have a good dispersion in the polymer matrix by using mechanical stirring, ultrasound or other dispersion methods.23 Molten ε-caprolactam (CL) monomer has the ability to dissolve different kinds of nanofillers, in situ blending via anionic polymerization of CL could be regarded as a more suitable way to prepare PA6 blends with excellent properties.24 Solid reactive extrusion (where the mixture is blended in solid state) has been examined as an effective approach to fabricate PA6/polymer blends, such as PA6/polypropylene and PA6/polyethylene,24 but, there are few reports about PA6/nanofillers composites (especially PA6/graphene) prepared by liquid reactive extrusion (the mixture is blended as a liquid).
In this work, we report an approach to fabricate PA6/graphene nanocomposites by liquid reactive extrusion. The graphene used in this work was few-layer graphene (FLG), due to the fact that single layer graphene is very difficult to obtain. FLG was synthesized by a high-volume production method and dispersed in molten ε-caprolactam. The morphology and mechanical properties of FLG/PA6 nanocomposites with different contents of FLG were investigated. It was found that liquid reactive extrusion was an effective method to prepare graphene/PA6 nanocomposites in industrial production.
2. Experimental
2.1 Materials
Expanded graphite powder was supplied by Qingdao Nanshu Hongda Graphite Products Co., Ltd. Commercial grade ε-caprolactam (CL, purity ≥ 99.0%) was obtained from Nanjing Oriental Chemical Co., Ltd. Semiconductor grade-sodium hydroxide (NaOH, 99.99% metal basis), ethylene glycol and ethanol were purchased from Sinopharm Chemical Reagent Co., Ltd. Sulfuric acid (H2SO4, 98.0%), hydrogen peroxide (H2O2, 30% aq.) and analysis purity toluene-2,4-diisocyanate (TDI, HPLC 99.5%) was purchased from Shanghai Chemical Reagents Co., Ltd. N-Methyl pyrrolidone (NMP, purity ≥ 99.0%) was supplied by Tianjin Guangfu Fine Chemical Research Institute.2.2 Preparation of few-layer graphene
A method to prepare large scale few-layer graphene has been reported by Fu et al. (2014),9,25 which we improved with a more time-saving process. To produce FLG, a certain amount of expanded graphite powder was added to ethanol, and the mixture was dispersed through an ultrasonic cell crusher (JY92-IIDN, 20–25 kHz, 900 W, Ningbo Scientz Co., Ltd, China) for 20 minutes. The solution was then filtered by vacuum filtration and dried at 70 °C for 10 hours in a vacuum oven, and the graphite nanoplates were obtained. Later, the graphite nanoplates were added to oleum, the mixture was sonicated for 30 minutes before filtrated and then 10% hydrogen peroxide was added and sonicated for 25 minutes. After that, the solution was filtered and washed with deionized water. The samples were dehydrated and dried in a vacuum oven at 80 °C for 10 hours, and the intercalated graphite nanoplates powder was obtained. Then, the intercalated graphite nanoplates powder was added to ethylene glycol through the re-expanded process to obtain re-expansion graphite nanoplates. After this, the re-expansion graphite nanoplates were added to N-methyl pyrrolidone and the solution was sonicated for about 3 hours the solution was later filtered and dried to obtain the graphene sheets. Finally, the graphene sheets powder was purified by protection (nitrogen) and reduction (hydrogen) mixed gas, and the FLG powder was obtained.2.3 Preparation of FLG/PA6 nanocomposites
A desired amount of FLG powder was added to molten ε-caprolactam, the mixture was sonicated with the ultrasonic cell crusher mentioned above. Then, the mixture was vacuumed at 140 °C for 30 minutes to remove trace water. After that, the molten mixture was cooled down to 120 °C and divided into two parts of equal weight. Each part was stored in a stirred tank under nitrogen, and then 5 wt% NaOH was added into one of the tank, and 0.5 wt% TDI was added into the other one. After dissolution with NaOH and TDI, the molten mixture was introduced into a twin-screw extruder from the two tanks by heated gear pumps with the same flow rate at 120 °C. The processing temperatures of the twin screw extruder are in the range of 120–240 °C, and the screw rotational speed was 200–300 rpm. The mixture was polymerized, extruded and granulated, and FLG/PA6 nanocomposites with different FLG content were obtained (details of these samples are given in the ESI, Fig. S1†). For a comparison, pure PA6 was also prepared by the same process. The master batches were dried for injection molding (Fig. S1†).3. Characterization
High resolution transmission electron microscopy (HR-TEM) was conducted using a 200 kV F20ST (FEI company).Raman spectroscopy measurements were obtained using a SPEX-1403 laser Raman spectrometer with excitation provided in back-scattering geometry by a 532 nm argon laser line.
Field-emission scanning electron microscopy (FE-SEM) images were taken on a Hitachi S4800 field-emission SEM system.
X-Ray photoelectron spectroscopy (XPS) measurements were performed in a VG ESCALB MK-II electron spectrometer, using a monochromatic Al Kα X-ray source operated at 1486 eV.
Fourier-transformed infrared (FT-IR) analysis was conducted using a Nicolet 6700 spectrophotometer (Nicolet Instrument Co., USA), the samples were prepared by co-compression of KBr powder with other samples.
A waters-991 gel permeation chromatography (GPC) instrument was used to evaluate the weight-average molecular weight (![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) w), the number-average molecular weight (n) and the polydispersities (w/n) of the PA6 composites. All of the samples were solved by m-cresol and FLG was filtered off.
w), the number-average molecular weight (n) and the polydispersities (w/n) of the PA6 composites. All of the samples were solved by m-cresol and FLG was filtered off.
X-Ray diffraction (XRD) patterns were employed to characterize FLG and FLG/PA6 composites using a Rigaku D/Max-Ra rotating anode X-ray diffractometer equipped with a Cu-Ka tube and Ni filter (λ = 0.1542 nm). The scan rate was 4°min−1. The FLG was performed as powder and the composites were performed as thin films (thickness: 3 mm).
The melting and crystallization behaviors of FLG/PA6 nanocomposites were characterized using a NETZSCH DSC 200 PC differential scanning calorimeter (DSC). The pristine PA6 and composites were first heated from 50 °C to 300 °C at a heating rate of 20°C min−1 under a nitrogen atmosphere and kept for 5 minutes to erase the thermal history. The crystallization behaviors were recorded from 300 °C to 50 °C at cooling rates of 20 °C min−1. Then the samples were second heated from 50 °C to 300 °C at a heating rate of 20 °C min−1. The melting temperature (Tm) and the degree of crystalline (Xc) (Xc = ΔH/ΔH100F, where ΔH is the enthalpy of fusion, ΔH100 is the enthalpy of fusion for a 100% crystalline PA6 which was taken to be 190 J g−1, the factor F denotes the fraction of polymer present in the composite.) were determined from the heating scan.
The tensile test of PA6 and FLG/PA6 nanocomposites was performed by using an Instron 1122 machine at room temperature, according to ASTM D 638 standard at a crosshead speed of 50 mm min−1. The flexural strength and flexural modulus were measured according to ASTM D 790.
4. Results and discussion
4.1 Structural and morphological characterizations of the synthesized few-layer graphene
TEM was used to determine the layers and quality of the FLG, Fig. 1A shows a wide field image for the FLG sheets with thick and wrinkled edges on the surface.26 The number of FLG sheets can be easily identified from their edges by using HR-TEM. As it can be seen from Fig. 1B–D, most of the FLG sheets contained 1 to 5 layers on average, corresponding to few-layer graphene.27 The thickness of a single layer, four layers and five layers was about 0.339, 1.37 and 1.71 nm, respectively, which corresponds with result obtained in the previous reports.25 Moreover, from the HR-TEM images, no obvious damages could be observed on the sheets, indicating that good quality FLG was obtained by our preparation method. | ||
| Fig. 1 HR-TEM images of the FLG powder prepared in this work: (A) is the wide field image of the FLG sheets, (B–D) are the images about the number layers of FLG. | ||
FLG was also characterized by Raman spectroscopy (Fig. 2), the G line (∼1585 cm−1) corresponds to the first-order scattering of E2g phonons, and 2D line (∼2701 cm−1) corresponds to the second-order of zone boundary phonons.26,28 Graphene can also be distinguished from less exfoliated and few-layered graphite by the intensity ratio between G and 2D peak (IG/I2D).26 From Fig. 2, the IG/I2D ratio decreased from 3.4 for graphite to 2.6 for FLG, implying the good exfoliation of graphite. Moreover, two apparent peaks were found in the 2D band of graphite, while there was only one main peak in the 2D band of FLG, indicating the high quality of the FLG obtained in this work. The apparent difference between graphite and FLG could be observed from the FE-SEM images too (as can be seen from Fig. S2, ESI†).
 | ||
| Fig. 2 Raman spectra of the graphite and the FLG powder. | ||
XPS offers direct evidence of the elemental composition and bonding of the graphene.12,29 As shown in Fig. 3A, the XPS survey scan spectrum of FLG shows two obvious peaks at 284.40 eV and 532.39 eV which can be attributed to C 1s and O 1s. The atomic composition of carbon and oxygen (97.81%, 2.19%) confirms that some oxygen still remained in the graphene, which is consistent with other reports.12,16,30 The C 1s XPS spectrum of graphene can be commonly attributed to five components, c1 (C-sp2), c2 (C-sp3), c3 (C–O), c4 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and c5 (π–π).31 Fig. 3B shows the high-resolution C 1s XPS spectrum of FLG, the peaks at 284.4, 285.3, 286.4 and 291.3 eV correspond to c1, c2, c3 and c5 components, respectively. The peaks for c3 and c5 were very weak, while the CO bond of c4 was difficult to be observed, implying good purity of FLG.
O) and c5 (π–π).31 Fig. 3B shows the high-resolution C 1s XPS spectrum of FLG, the peaks at 284.4, 285.3, 286.4 and 291.3 eV correspond to c1, c2, c3 and c5 components, respectively. The peaks for c3 and c5 were very weak, while the CO bond of c4 was difficult to be observed, implying good purity of FLG.
 | ||
| Fig. 3 XPS spectra of the FLG, where (A) shows general spectra and (B) shows the C 1s core-level spectra. | ||
4.2 Structural and morphological characterizations of FLG/PA6 nanocomposites
The dispersion and interfacial interaction of nanofillers and matrix are two important factors influencing the macroscopic properties of composites.32–34 In order to understand the dispersion state of the FLG nanoplates in the PA6 matrix, fractured surfaces of PA6 and FLG/PA6 nanocomposites with 0.5 wt% FLG were investigated by SEM. As shown in Fig. 4B, several FLG sheets could be observed on the fractured surface and they were relatively well dispersed throughout the PA6 matrix as pointed by the yellow circles, while neat PA6 exhibited a typical smooth morphology (Fig. 4A). Fig. 4C and D shows images with a high magnification of the regions shown in red in Fig. 4B. It was clearly found that the interfacial interaction between the FLG sheets and the PA6 matrix were adhered well, similar to the reported interaction between graphene and other polymers.35 This might be potentially due to the formation of strong hydrogen bonds between the FLG sheets and the polar PA6 molecular chains.34,35 Which could be further confirmed by the analysis of FTIR and Raman spectrum in the below discussion (the dispersion process of FLG in PA6 matrix is shown in Fig. S3, ESI†). | ||
| Fig. 4 FE-SEM images of the PA6 and the FLG/PA6 composites, where (A) shows the image of the pure PA6 and (B) shows the image of the FLG/PA6 composites with 0.5 wt% FLG; (C) and (D) show the high magnification images of points a and b of (B). | ||
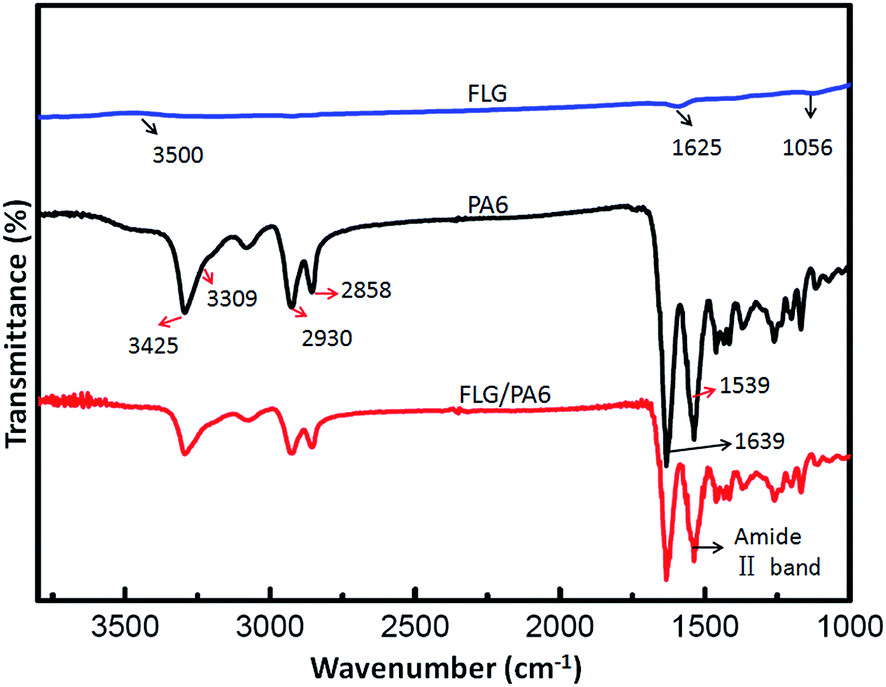
FTIR characterizations of FLG, PA6 and FLG/PA6 nanocomposite were performed to observe the functional groups. As can be seen from Fig. 5, in the FTIR spectrum of FLG, the peaks at 1056 and 1625 cm−1 are correspond to the C–O (alkoxy) and CC stretching vibration, respectively; the peak around 3500 cm−1 which is attributed to stretching of the O–H band is very unobvious, indicating that the sample with low water content; the peak at about 1720 cm−1 (corresponding to the CO) is almost can't be found, which is consistent with the result of the above XPS spectra. For the FTIR spectrum of pure PA6, the peak at 1639 cm−1 is corresponds to the CO stretching; the peak at 1539 cm−1 is due to the C–N stretching; the peaks at 2858 and 2930 cm−1 are correspond to the C–H stretching; the ε-caprolactam has been polymerized to PA6 was suggested by the peaks at 3309 cm−1 for N–H and 3425 cm−1 for O–H, respectively.17,30 Compared to the pure PA6, there are no apparent changes in the FTIR spectrum of FLG/PA6 nanocomposite, which might be because of the low concentration of FLG. The FLG/PA6 nanocomposite almost has all the absorption peaks of pure PA6. However, the peaks' intensity of PA6 becomes increasingly weak with the FLG content increased. When FLG was added to PA6, the amide II band (at about 1536 cm−1) became less intense and subtly shifted, which suggested that FLG has been interacted with PA6 by hydrogen bonding.36 From Fig. 5, the weaker bands of asymmetric vibration at the area between 3000 and 3300 cm−1 stretching in the spectrum of FLG/PA6 nanocomposite were found, which revealed that the good dispersion of FLG sheets in the PA6 matrix can destroy the intermolecular hydrogen bonds of PA6 chains, and form the new hydrogen bonds between FLG sheets and PA6 matrix.36
 | ||
| Fig. 5 FTIR spectra of FLG, pure PA6 and FLG/PA6 nanocomposite (with 1 wt% FLG). | ||
The interaction between FLG and PA6 matrix can also be demonstrated by Raman spectroscopy. Fig. 6 shows the Raman spectrum of FLG and FLG/PA6 nanocomposite, the three characteristic peaks at around 1352 cm−1 (D line), 1585 cm−1 (G line) and 2701 cm−1 (2D line) of FLG can still be found in the Raman spectrum of FLG/PA6 nanocomposite with the same position. However, a clear intensity decrease in G and 2D peak of FLG/PA6 nanocomposite can be observed when compared to that of the pure FLG, indicating the existence of strong interaction between FLG and PA6 matrix. Consideration the results of the FTIR, this interaction force may be attributed to the hydrogen bonds. Moreover, the intensity ratio between D and G peak (ID/IG) increase from ∼0.05 in FLG to ∼0.18 in FLG/PA6 nanocomposite, implying the further reduction of FLG during the polymerization process of PA6, similar results can be found in other reported works.30,37,38 It has been confirmed that the molten ε-caprolactam can act as a reducing agent,39,40 so, the remaining oxygen containing groups of the FLG may be reduced during the polymerization process, thus resulted in the high quality of FLG sheets in the nanocomposites, and caused the apparent changes of ID/IG.
 | ||
| Fig. 6 Raman spectra of FLG and FLG/PA6 nanocomposite. | ||
4.3 Molecular weight and monomer conversion analysis
Table 1 lists thew, n and w/n for the PA6 and the FLG/PA6 nanocomposites, as well as the monomer conversion data [monomer conversion percentage = (polymer weight after extraction)/(polymer weight before extraction)]. It can be seen that the w, n and the monomer conversion of the PA6 composites showed little changed when the content of FLG increased. These results indicate that the incorporation of the FLG had no obvious effect on the molecule weight and the monomer conversion of the PA6 composites.
| Samples | w |
n |
w/n |
Conversion (%) |
|---|---|---|---|---|
| PA6 | ∼49800 |
∼21700 |
2.29 | 99.32 |
| FLG/PA6 0.1 | ∼49600 |
∼21200 |
2.34 | 98.52 |
| FLG/PA6 0.5 | ∼48200 |
∼20600 |
2.34 | 99.21 |
| FLG/PA6 1 | ∼48100 |
∼20500 |
2.35 | 98.47 |
4.4 Crystalline properties
The crystalline structures of the FLG and the FLG/PA6 were assessed by using XRD. As shown in Fig. 7, the diffraction peaks located at about 2θ = 20.1°and 24° correspond to (200) and (002, 202) reflections of the α-form crystals of the PA6, respectively.17,41,42 When 0.5 wt% FLG was incorporated, the representative diffraction peak located at 2θ = 26.2°was found, which can be attributed to the (002) crystalline plane of the graphene.43 When the FLG content increased to 1 wt%, the diffraction peak of the graphene became clear, indicating that the FLG sheets were well dispersed in the PA6 matrix. No changes in the typical structures were produced, while some changes were found in the intensity of α1 when the FLG loading increased, implying that the crystallization behavior of the polymers will be affected when nanofillers are added.44,45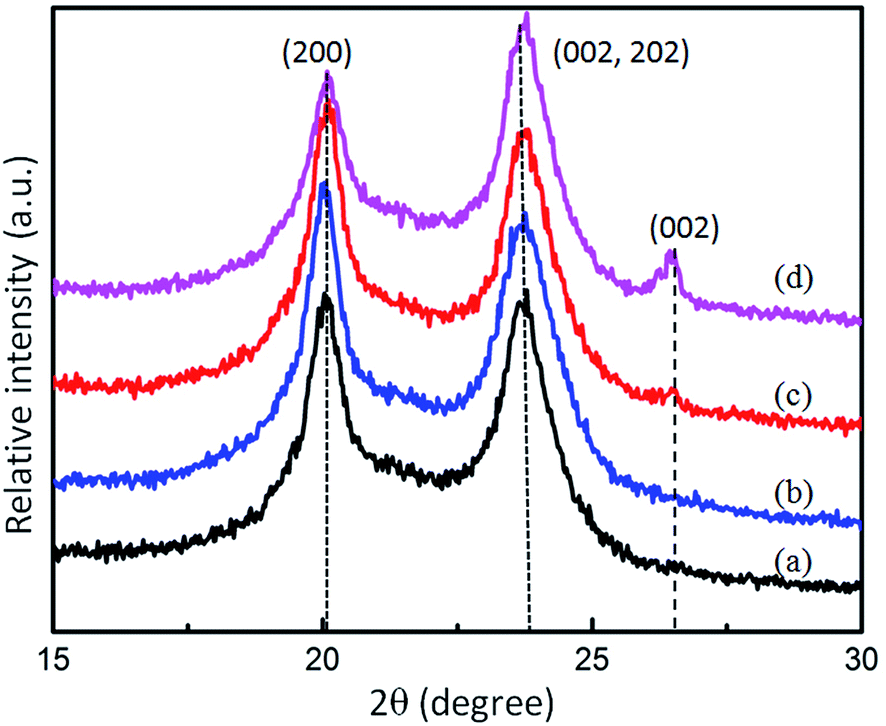 | ||
| Fig. 7 XRD patterns of the pure PA6 and FLG/PA6 nanocomposites with various loading of FLG (from bottom to top): (a) PA6; (b) FLG/PA6 0.1; (c) FLG/PA6 0.5; (d) FLG/PA6 1.0. The curves are vertically offset for clarity. | ||
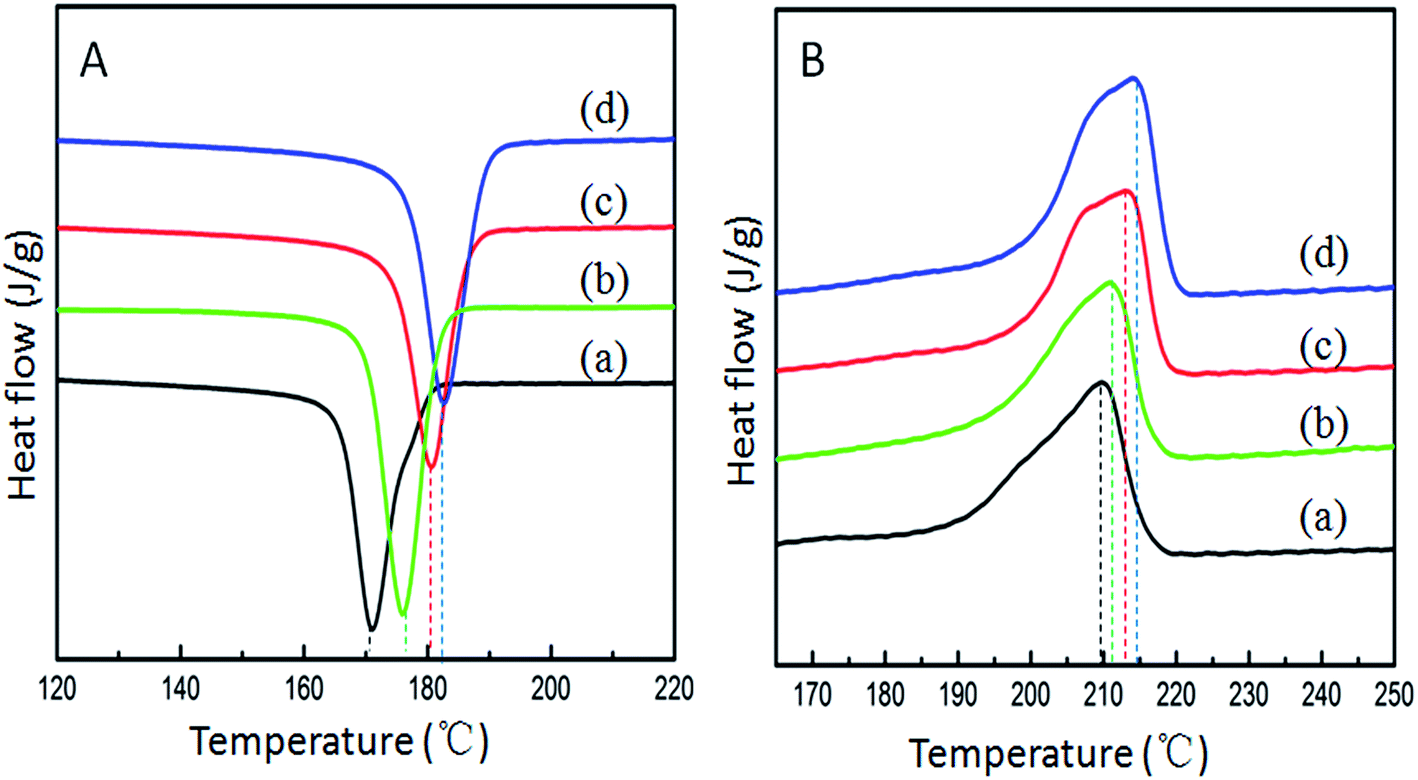
The effect of FLG on the melt and crystallization behavior of the pure PA6 and FLG/PA6 nanocomposites were also investigated by DSC. The cooling second heating curves are shown in Fig. 8, and the data is summarized in Table 2. It can be seen from Fig. 8A that the incorporation of FLG led to an obvious increase in the crystallization temperature (Tc), indicating that the FLG acted as a nucleation agent during the crystallization of the PA6. It is well known that PA6 has three crystals forms: the more thermodynamically stable α-form, γ-form and the unstable β form.17 Fig. 8B presents the DSC second heating scans of the PA6 and the composites. It can be observed that there was one apparent melting peak and an obscure shoulder peak in the PA6 and the composites spectra. As the γ-form was not found in the XRD patterns, the peaks found here should correspond to the α-form crystal.
 | ||
| Fig. 8 DSC cooling scan (A) and second heating scan (B) of the PA6 and the FLG/PA6 nanocomposites (from bottom to top): (a) PA6; (b) FLG/PA6 0.1; (c) FLG/PA6 0.5; (d) FLG/PA6 1.0. The curves are vertically offset for clarity. | ||
| Samples | Tm (°C) | ΔHm (J g−1) | Xc (%) | Tc (°C) |
|---|---|---|---|---|
| PA6 | 209.84 | 48.58 | 25.57 | 171.03 |
| FLG/PA6 0.1 | 211.06 | 50.50 | 26.58 | 175.88 |
| FLG/PA6 0.5 | 213.18 | 51.52 | 27.12 | 180.66 |
| FLG/PA6 1 | 214.17 | 60.83 | 32.02 | 182.64 |
From the data listed in Table 2, it can be seen that the melting temperature (Tm), crystallization temperature (Tc) and the crystallinity (Xc) of the nanocomposites increased in comparison with that of the pure PA6, further confirming that the well dispersed FLG sheets can promote the crystallization by acting as a nucleating agent.
4.5 Mechanical properties
An improved interfacial interaction between the nanosheets and the matrix in the FLG/PA6 nanocomposites has been observed by the SEM and the XRD data shown earlier. A considerable reinforcement in the mechanical properties of the PA6 can be expected by incorporating FLG sheets into the PA6 matrix.41 The tensile and flexural samples of FLG/PA6 nanocomposites and pure PA6 were prepared by injection molding. It can be seen in Fig. 9 that the tensile and flexural strength of the nanocomposites showed a trend of first increasing and then decreasing. By including 0.1 and 0.5 wt% FLG, the tensile and flexural strength increased ∼10.08%, ∼12.66% and ∼18.95%, ∼42.99%, respectively (Fig. 9A and B). When 1 wt% FLG was added the tensile and flexural strength decreased. The increase in the strength of the composites with low FLG content might be because of the superior mechanical properties showed by the FLG sheets or their high surface area and the good dispersion of FLG sheets in the PA6 matrix.9,46 When the loading of FLG continuously increased, a reduction of strength was observed, which has also been observed in previously reported work where graphene-based nanofillers were used.47–51 The lower strength at high graphene content can be ascribed to the follow reasons:52 (1) owing to the π–π interactions between graphene sheets and strong van der Waals forces, FLG sheets tending to aggregating when its content reaching to a relative higher level; (2) the aggregated FLG sheets may act as micrometer-size fillers with very smaller interface area, so, the mechanical strength of FLG will diminish gradually; (3) the uncoiling and straightening of polymer macromolecules will be restricted because of the steric hindrance made by the aggregated FLG at high portions; (4) the relative big aggregations of FLG may act as stress-concentration points in the nanocomposites and resulted in the bad strength of the nanocomposites.53 This also can be approved by the FE-SEM images of nanocomposites with high volume of graphene in Fig. S4 and S5 (ESI†).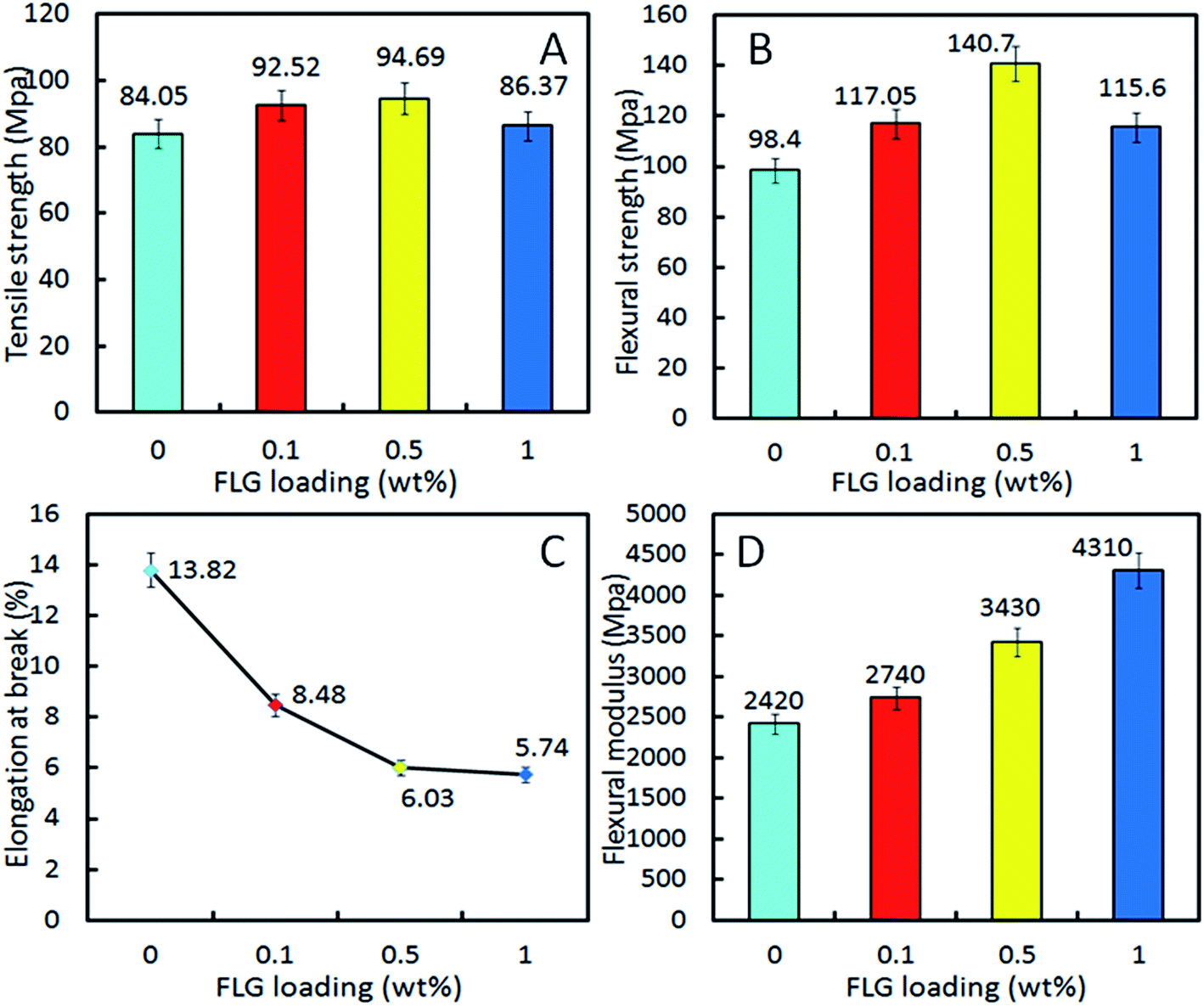 | ||
| Fig. 9 Mechanical properties testing: (A) tensile strength; (B) flexural strength; (C) elongation at break; (D) flexural modulus. | ||
Generally speaking, the elongation of composites will be reduced when fillers with higher stiffness than the matrix.53 It is apparent that the stiffness of graphene is higher than that of the PA6 matrix, so, a clear tendency for a decreasing in elongation at break with the increasing amount of FLG has been observed. As can be seen from Fig. 9C, the average values of elongation at break decreased to ~5.74% for FLG/PA6 nanocomposite with 1 wt% FLG from ~13.82% for pure PA6. The reason can be attributed to the improved interfacial interaction between the graphene and matrix which reduced the PA6 chain mobility.49
Fig. 9D shows the flexural modulus results of pure PA6 and the nanocomposites. An obvious increase of the modulus was observed comparing to pure PA6. When incorporating 0.1, 0.5 and 1 wt% FLG, the flexural modulus was enhanced by ∼13.22%, ∼41.74% and ∼78.10%, respectively. Clearly, this trend of the modulus was different than that observed for the flexural strength, which may be attributed to the factors like these: with the FLG loading increased, the toughness of the nanocomposites decreased, thus improved the ability to resist the flexural deformation of nanocomposites, and resulted in the increase of the flexural modulus. While the measurement standard on the modulus and strength is different, the flexural strength is main related to the tensile and compression resistance. Therefore, it is not strange to find the different trend between the flexural strength and the flexural modulus.
5. Conclusions
High quality few-layer graphene was produced using a re-expansion and exfoliation method. Then, FLG/PA6 nanocomposites were prepared through a liquid reactive extrusion process. FE-SEM results demonstrated that the FLG sheets were well dispersed in the PA6 matrix. An obvious reinforcement of the mechanical properties of the nanocomposites were obtained by incorporating low contents of FLG, the tensile and flexural strength of the nanocomposites with 0.5 wt% FLG loading were enhanced by ∼12.66% and ∼42.99%, respectively, compared to that of the pure PA6. The flexural modulus of the nanocomposites was improved by ∼78.10% when 1 wt% FLG was added. Moreover, incorporating low content of FLG could improve the crystallization rate and the crystallization temperature of the nanocomposites as the graphene can act as a nucleate. FLG/PA6 nanocomposites were successfully fabricated by liquid reactive extrusion, and it has been showed that liquid reactive extrusion was a continuous, facile and effective method to prepare graphene/polymer nanocomposites. Therefore, this approach will lead to the commercial production of graphene/polymer nanocomposites with low content of nanofillers and with satisfactory mechanical properties.Acknowledgements
We would like to extend our gratitude to Hefei Genius Advanced Material Co., Ltd for the use of their facilities and equipment. We also gratefully acknowledge the financial support from the National Natural Science Foundation of China (2013BAE02B03).References
- G. Rajni, K. D. Naba and R. C. Namita, Nanomaterials, 2014, 4, 267–300 CrossRef PubMed.
- R. M. Hazen, R. T. Downs and A. Jones, Rev. Mineral. Geochem., 2013, 75, 7–46 CrossRef CAS.
- A. R. Oganov, R. J. Hemley and R. M. Hazen, Rev. Mineral. Geochem., 2013, 75, 47–77 CrossRef CAS.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich and S. V. Morozov, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451 CrossRef CAS PubMed.
- S. Hong-Kyu and L. Tae-Woo, Carbon Lett., 2013, 3, 145–151 Search PubMed.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- E. H. Hwang, S. Adam and D. Dassarma, Phys. Rev. Lett., 2007, 98, 186806 CrossRef CAS.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg and J. Hone, Solid State Commun., 2008, 146, 351–355 CrossRef CAS PubMed.
- C. Lee, X. D. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- L. Y. Ma, B. Yu, X. D. Qian, W. Yang, H. F. Pan and Y. Q. Shi, Polym. Adv. Technol., 2014, 25, 605–612 CrossRef CAS PubMed.
- Z. Y. Sun, P. Sascha, X. Huang, G. Dmitrii, T. Christoph and E. Petra, Carbon, 2013, 64, 288–294 CrossRef CAS PubMed.
- C. L. Bao, L. Song, W. Y. Xing, B. H. Yuan, C. A. Wilkie and J. L. Huang, J. Mater. Chem., 2012, 22, 6088–6096 RSC.
- B. Ahmadi-Moghadam and F. Taheri, J. Mater. Sci., 2014, 49, 6180–6190 CrossRef CAS.
- X. B. Fu, C. G. Yao and G. S. Yang, RSC Adv., 2015, 5, 61688–61702 RSC.
- D. Verma, P. C. Gope, A. Shandilya and A. Gupta, Trans. Indian Inst. Met., 2014, 67, 803–816 CrossRef CAS.
- A. O'Neill, D. Bakirtzis and D. Dixon, Eur. Polym. J., 2014, 59, 353–362 CrossRef PubMed.
- P. Ding, S. S. Su, N. Song, S. F. Tang, Y. M. Liu and L. Y. Shi, Carbon, 2014, 66, 576–584 CrossRef CAS PubMed.
- E. Tkalya, M. Ghislandi, A. Alekseev, C. Koning and J. Loos, J. Mater. Chem., 2010, 20, 3035–3039 RSC.
- M. I. Oana, R. P. Keith, K. Vmar, O. Arlene, P. B. Alan and N. C. Jonathan, Carbon, 2014, 78, 243–249 CrossRef PubMed.
- V. Raquel, B. M. Mar, J. R. Laura and A. L. M. Miguel, J. Mater. Chem., 2011, 21, 3301–3310 RSC.
- T. Villmow, P. Potschke, S. Pegel, L. Haussler and B. Kretzschmar, Polymer, 2008, 49, 3500–3509 CrossRef CAS PubMed.
- M. T. Muller, B. Krause, B. Kretz Schmar and P. Potschke, Compos. Sci. Technol., 2011, 71, 1535–1542 CrossRef PubMed.
- H. Sven, S. Robert, P. Doris, V. Brigitte, H. Kathrin and P. Petra, Compos. Sci. Technol., 2012, 72, 1671–1677 CrossRef PubMed.
- D. G. Yan, G. Li, M. Y. Huang and C. Wang, Polym. Eng. Sci., 2013, 53, 2705–2710 CAS.
- Y. X. Fu, X. M. Wang, D. C. Mo and S. S. Lu, J. Mater. Sci., 2014, 49, 2315–2323 CrossRef CAS.
- S. Castarlenas, C. Rubio, A. Mayoral, C. Tellez and J. Coronas, Carbon, 2014, 73, 99–105 CrossRef CAS PubMed.
- V. Chabot, B. Kim, B. Sloper, C. Tzoganakis and A. Yu, Sci. Rep., 2013, 3, 1378 Search PubMed.
- C. T. J. Low, F. C. Walsh, M. H. Chakrabarti, M. A. Hashim and M. A. Hussain, Carbon, 2013, 54, 1–21 CrossRef CAS PubMed.
- X. Q. Zhang, X. Y. Fan, H. Z. Li and C. Yan, J. Mater. Chem., 2012, 22, 24081–24091 RSC.
- P. Ding, S. S. Su, N. Song, S. F. Tang, Y. M. Liu and L. Y. Shi, RSC Adv., 2014, 4, 18782–18791 RSC.
- X. Q. Liang, J. Zhong, Y. L. Shi, J. Guo, G. L. Huang and G. H. Hong, Mater. Res. Bull., 2015, 61, 252–258 CrossRef CAS PubMed.
- S. Gayathri, P. Jayabal, M. Kottaisamy and V. Ramakrishnan, AIP Adv., 2014, 4, 027116 CrossRef PubMed.
- P. Ramalingam, S. T. Pusuluri, S. Periyasamy, R. Veerabahu and J. Kulandaivel, RSC Adv., 2013, 3, 2369–2378 RSC.
- N. N. Hong, L. Song, T. R. Hull, A. A. Stec, B. B. Wang and Y. Pan, Mater. Chem. Phys., 2013, 142, 531–538 CrossRef CAS PubMed.
- Z. Fan, F. Gong, S. T. Nguyen and H. M. Duong, Carbon, 2015, 81, 396–404 CrossRef CAS PubMed.
- H. R. Pant, B. Pant, P. Pokharel, H. J. Kim, L. D. Tijing, C. H. Park, D. S. Lee, H. Y. Kim and C. S. Kim, J. Membr. Sci., 2013, 429, 225–234 CrossRef CAS PubMed.
- W. Li, B. Q. Zhou, M. Y. Wang, Z. H. Li and R. Ren, J. Mater. Sci., 2015, 50, 5402–5410 CrossRef CAS.
- H. H. Liu, W. W. Peng, L. C. Hou, X. C. Wang and X. X. Zhang, Compos. Sci. Technol., 2013, 81, 61–68 CrossRef CAS PubMed.
- J. P. Duan, D. J. Zhao and G. S. Yang, RSC Adv., 2014, 4, 28765–28768 RSC.
- Z. Xu and C. Gao, Macromolecules, 2010, 43, 6716–6723 CrossRef CAS.
- L. Gong, B. Yin, L. P. Li and M. B. Yang, Composites, Part B, 2015, 73, 49–56 CrossRef CAS PubMed.
- C. Ramesh and E. B. Cowd, Macromolecules, 2001, 34, 3308–3313 CrossRef CAS.
- G. X. Wang, J. Yang, J. S. Park, X. L. Gou, B. Wang and H. Liu, J. Phys. Chem. C, 2008, 112, 8192–8195 CAS.
- B. Y. Li, H. H. Yuan and Y. Z. Zhang, Compos. Sci. Technol., 2013, 89, 134–141 CrossRef CAS PubMed.
- L. Sun, J. T. Yang, G. Y. Lin and M. Q. Zhong, Mater. Lett., 2007, 61, 3963–3966 CrossRef CAS PubMed.
- D. Dorian, L. Patrick, H. Jeremy, L. Genady and A. Edward, J. Thermoplast. Compos. Mater., 2013, 8, 1–18 Search PubMed.
- X. Zhao, Q. Zhang, D. Chen and P. Lu, Macromolecules, 2010, 43, 2357–2363 CrossRef CAS.
- P. Song, Z. Cao, Y. Cai, L. Zhao, Z. Fang and S. Fu, Polymer, 2011, 52, 4001–4010 CrossRef CAS PubMed.
- P. Gogoi, R. Boruah and S. K. Dolui, Prog. Org. Coat., 2015, 84, 128–138 CrossRef CAS PubMed.
- S. Paszkiewicz, A. Szymczyk, K. Livanov, H. D. Wagner and Z. Roslaniec, eXPRESS Polym. Lett., 2015, 9, 509–524 CrossRef CAS.
- L. D. Liu, C. Y. Tong, Y. He, Y. X. Zhao and C. L. Lu, J. Membr. Sci., 2015, 487, 99–108 CrossRef CAS PubMed.
- S. Araby, N. Saber, X. Ma, N. Kawashima, H. L. Kang, H. Shen, L. Q. Zhang, J. Xu, P. Majewski and J. Ma, Mater. Des., 2015, 65, 690–699 CrossRef CAS PubMed.
- N. Loan, S. M. Choi, D. H. Kim, N. K. Kong, P. J. Jung and S. Y. Park, Macromol. Res., 2014, 22, 257–263 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra14067f |
| This journal is © The Royal Society of Chemistry 2015 |