
Eutectic ionic liquid mixtures and their effect on CO2 solubility and conductivity†
Anna S. Ivanovaa,
Thomas Brinzerb,
Elliot A. Rothd,
Victor A. Kusumad,
John D. Watkinsd,
Xu Zhoudc,
David Luebked,
David Hopkinsond,
Newell R. Washburna,
Sean Garrett-Roe *b and
Hunaid B. Nulwala*ad
*b and
Hunaid B. Nulwala*ad
aDepartment of Chemistry, Carnegie Mellon University, 4400 Fifth Avenue, Pittsburgh, PA 15213, USA. E-mail: hnulwala@andrew.cmu.edu
bUniversity of Pittsburgh, Department of Chemistry, Chevron Science Center, 219 Parkman Avenue, Pittsburgh, PA 15260, USA. E-mail: sgr@pitt.edu
cLiquid Ion Solutions, LLC, 1817 Parkway View Drive, Pittsburgh, PA 15205, USA
dNational Energy Technology Laboratory, P.O. Box 10940, Pittsburgh, Pennsylvania 15236, USA
First published on 26th May 2015
Abstract
A simple binary system of compounds resembling short-chain versions of popular ionic liquids has been shown to have surprisingly complex properties. Combining methylated versions of pyridinium and pyrrolidinium bis[(trifluoromethyl)sulfonyl]imide gave desirable properties such as low viscosity and high conductivity solubility per unit volume. The binary combinations studied in this study showed that these materials were stable liquids at 50 °C and had a threefold improvement in conductivity over [C6C1im][Tf2N]. Despite the high densities of these materials, 2D-IR studies indicate increased ion mobility, likely due to the lack of hindering alkyl chains.
Introduction
Ionic liquids (ILs) have a potential advantage over existing solvents as a green medium due to their wide liquid range, outstanding solvation potential, negligible vapour pressure, thermal stability, and recyclability.1–3 These properties have prompted their use as lubricants, electrolytes for energy storage, CO2 capture media, coatings, and fuel cells.4–7 However, the optimization of IL properties for a given application is a challenge. Through skilful manipulation of the IL structures, one can target specific properties such as viscosity, solubility, conductivity, melting point, density, and refractive index.6,8–12 In principle, one can design novel ILs with the exact properties needed, but in practice, this is a daunting goal to achieve.ILs are complex solvents, whose properties ultimately depend on multiple intermolecular forces, including hydrogen bonding, ionic/charge–charge, dipolar, π–π, n–π, and van der Waals interactions. These interactions are quite complex when compared to simple solvents. Thus, it is difficult to tune the properties of ILs without extensive synthetic manipulation and careful characterization. One simple, alternative route to access specific properties is by creating mixtures of ILs.13,14 The properties of IL mixtures have been shown to be distinct from those of the parent compounds and researchers have started to evaluate binary and ternary mixtures.15–24 The mixing of ILs can improve liquid range and compound various properties of the ILs. Ionic materials having melting points over 100 °C have not been extensively studied and there remains an opportunity to further extend their useful properties.23 The exception is the work of Maximo et al.23 who have studied the melting behaviour of ionic solids mixtures.
Ionic solids are crystalline materials with high density. Similar to normal salts, these organic solids show eutectic behaviour upon mixing. In our work, various compositions of mixtures combining 1,1-dimethylpyrrolidinium, 1-methylpyridinium and bis[(trifluoromethyl)sulfonyl]imide (which are crystalline solids at room temperature) ([C1C1pyrr]x[C1pyr](1−x)[Tf2N]) (subscripts indicate the mole fraction) (Fig. 1) were studied. This combination is unusual because it lacks the bulky alkyl chains normally used to disrupt the crystal lattice packing and lower the melting points of traditional ILs. In contrast to Maximo's work, the anion used in this study is elongated.
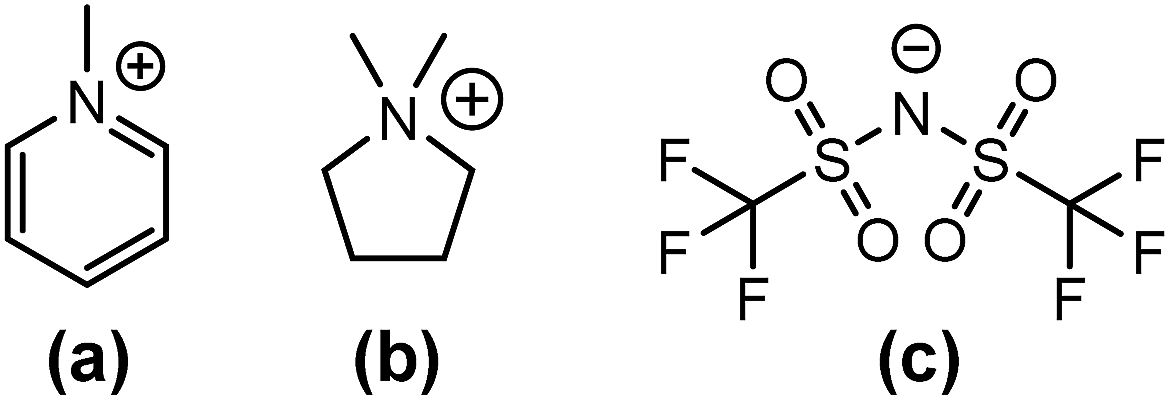 | ||
| Fig. 1 Components of the synthesized compounds: (a) 1-methylpyridinium cation [C1pyr]; (b) 1,1-dimethylpyrrolidinium cation [C1C1pyrr]; and (c) bis[(trifluoromethyl)sulfonyl]imide anion [Tf2N]. | ||
Typically, ILs rely on molecular asymmetry to disrupt the crystalline ordering of the ions and widen the liquid range.25 With symmetric cations, this effect must be achieved by other means. By combining two cations with different electronic structures or crystal phases, we aimed to introduce defects into the molecular ordering and induce a eutectic effect, resulting in a lower melting point.14 In addition, the shortened alkyl chains should eliminate the problem of tail group domains causing spatial heterogeneity,26 potentially increasing the rate of diffusion through the IL and thus improving the kinetics of gas adsorption.27 Though gas solubility is known to generally increase with the size and flexibility of ionic structures, for CO2 the dominant effect is the gas–anion interaction.28 Finally, the decreased cation size should increase the charge density of the mixture, possibly improving the conductivity. Thus, the combination [C1C1pyrr]x[C1pyr](1−x)[Tf2N] should result in a low-melting, conductive mixture with good gas solubility, making it widely applicable.
Experimental
Synthetic details
Note: all compounds were tested with aq. silver nitrate (1 M) solution. No visible silver halide formation was observed.
Experimental details are provided in the ESI.†
Results and discussion
Melting behaviour
The melting point and thermal stability of the mixtures were used as an initial screening methodology. The thermal stability was determined using thermogravimetric analysis (TGA) to identify the onset temperature of decomposition for each sample. In all cases, the onset point was above 415 °C with a ramp rate of 10 °C min−1, representing a ∼65 °C improvement over typical IL decomposition temperatures. This improvement was due to the removal of alkyl chains, which would normally decompose before the rest of the molecule, mainly due to the presence of alpha and beta protons. The absence of the alkyl chain lengths would also result in higher ionicity and conductivity. The melting points were obtained by differential scanning calorimetry (DSC) using a 10 °C min−1 ramp rate and were plotted to observe the effect of the sample composition on the melting temperature (Fig. 2). We also performed modulated DSC experiments on 3 samples and found no deviation from normal DSC in this case. In cases where multiple peaks were observed, the highest-temperature peak was assumed to be the melting point, ensuring a fully liquid sample. Nevertheless, the multi-phasic behaviour of such samples indicates the potential plastic29 or liquid crystal30 properties and is worth further exploration for electrolytic applications. The lowest melting point of the combinations was observed for [C1C1pyrr]0.3[C1pyr]0.7[Tf2N], which exhibits a shallow eutectic point of 33.0 °C. In total, four samples ranging from 0%–30% [C1C1pyrr][Tf2N] were completely liquid below 50 °C. We focused on analysing the CO2 capture properties of these low-melting candidates.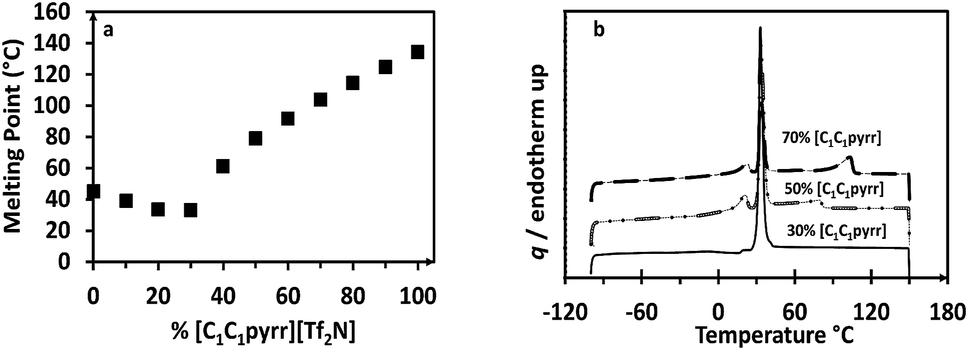 | ||
| Fig. 2 (a) DSC heating curves for [C1C1pyrr]0.3[C1pyr]0.7[Tf2N] (solid), [C1C1pyrr]0.5[C1pyr]0.5[Tf2N] (short dashes), and [C1C1pyrr]0.7[C1pyr]0.3[Tf2N] (long dashes). (b) Melting points for the binary combinations of ILs measured using DSC with a heating rate of 10 °C min−1. | ||
Interestingly, the pyridinium parent compound and several of the binary combinations exhibit a strong supercooling effect, with crystallization occurring at as much as 60 °C below the melting temperature. These samples were meta-stable liquids at room temperature unless crystallization was seeded or initiated through mechanical forces, at which point rapid crystallization was observed. Such behaviour suggests a kinetically hindered nucleation step in the pyridinium compound, possibly exacerbated in the mixtures due to the complex stacking interactions of multiple cations.
Viscosity
Low viscosity of the IL is responsible for enhancing the kinetics in gas capture and reaction media. Viscosities were measured at 50 °C for the low-melting samples (Table 1). 1-Hexyl-3-methylimidazolium bis[(trifluoromethyl)sulfonyl]imide or [C6C1im][Tf2N] was used as a baseline standard for the efficacy of other ILs20,31,32 and has been selected as a reference ionic liquid for an IUPAC experimental validation project. It has a melting point of −7 °C.18 At 50 °C, the viscosity of [C6C1im][Tf2N] is 26 mPa s, which is higher than most of our low-melting candidates at 50 °C. Though the melting temperature steadily decreases from 0%–30% [C1C1pyrr][Tf2N], the viscosity increases along the series. This unexpected trend suggests that the individual components are more important in determining the viscosity than the proximity of measuring temperature to the melting point.| [C6C1im][Tf2N] | [C1pyr][Tf2N] | % [C1C1pyrr][Tf2N] | |||
|---|---|---|---|---|---|
| 10 | 20 | 30 | |||
| a Measured for liquid at 50 °C and ∼1 atm. | |||||
| Tm, °C | −7 (ref. 9) | 45 | 39 | 34 | 33 |
| ηa, mPa s | 26 (ref. 9) | 23.9 | 24.7 | 25.1 | 26.2 |
| ρa, g cm−3 | 1.35 (ref. 11) | 1.58 | 1.57 | 1.55 | 1.54 |
| kHa (atm) | 46.8 | 66.5 | 52.3 | 47.6 | 51.7 |
| κa (mS cm−1) | 4.07 | — | 11.2 | 12.2 | 11.9 |
| Cpa (J mol−1 K−1) | 604 (ref. 18) | 434 | 454 | 462 | 463 |
CO2 uptake and density
The densities of the samples at 50 °C are all significantly above those of [C6C1im][Tf2N], as expected for a system involving small, symmetrical cations and lacking alkyl chains (Table 1). A slight decrease in density as the fraction of [C1C1pyrr][Tf2N] increases was noted, which can be explained by the increased proportion of pyrrolidinium cations interfering with the orderly stacking.Intuitively, a high density should lower free volume and reduce CO2 uptake.33 We find, however, that increase in ionic concentration improves the CO2 solubility of the overall system per unit volume. The CO2 solubility of the IL mixtures was evaluated by equilibrating each liquid under varying pressures of CO2 gas at 50 °C and measuring the amount absorbed. These results were then fitted to a polynomial curve and interpolated to account for the variance in pressure during the measurements. The results indicate that on the practical basis of moles of CO2 per volume of IL mixture, the [C1C1pyrr]0.2[C1pyr]0.8[Tf2N] showed comparable absorbance of CO2 to [C6C1im][Tf2N] at low pressures and up to 8% higher at elevated pressures (Fig. 3), which makes it more applicable for warm gas purification. It is suspected short alkyl chains increase the overall ionic nature of the system, improving the CO2 solubility per unit volume. Such increased CO2 solubility is promising for future trials with mixed systems of truncated cations.
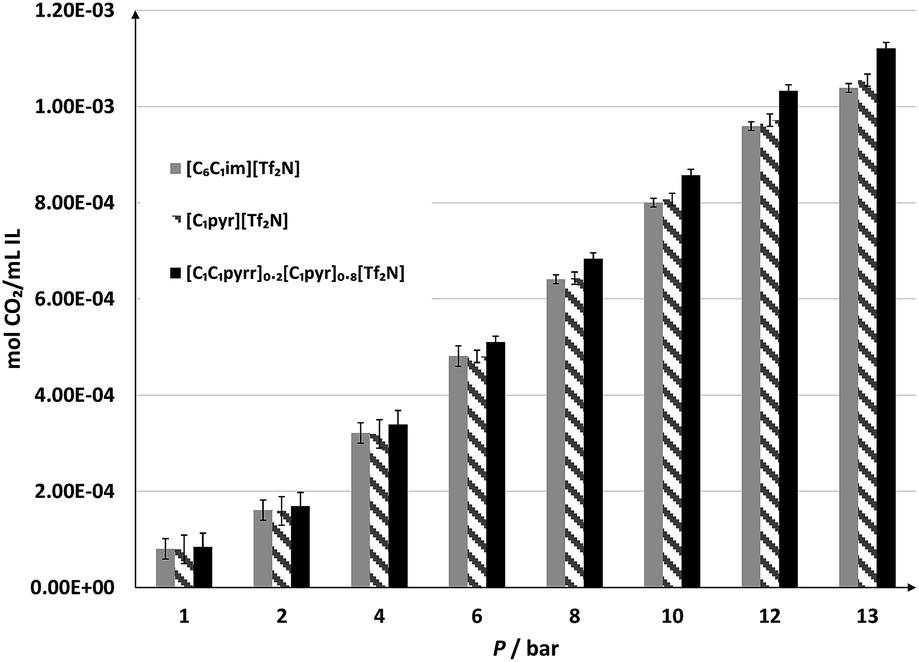 | ||
| Fig. 3 CO2 sorption of [C6C1im][Tf2N],34 [C1pyr][Tf2N] and the best-absorbing binary mixture measured at 50 °C and various pressures, interpolated from the experimental data. Sorption is shown as moles CO2 absorbed by a given starting volume of ionic liquid. | ||
Heat capacity
For applications involving heating the ionic liquid, the heat capacity determines how much energy will be required. The heat capacity for these samples was measured using DSC at three different temperatures (Fig. 4). The heat capacities range from 430 to 621 J mol−1 K−1, with some fluctuation in the general upwards trend with increasing [C1C1pyrr][Tf2N] content. Overall, the heat capacities are lower than that of [C6C1im][Tf2N] most likely due to the fewer intramolecular degrees of freedom in the smaller ionic mixtures.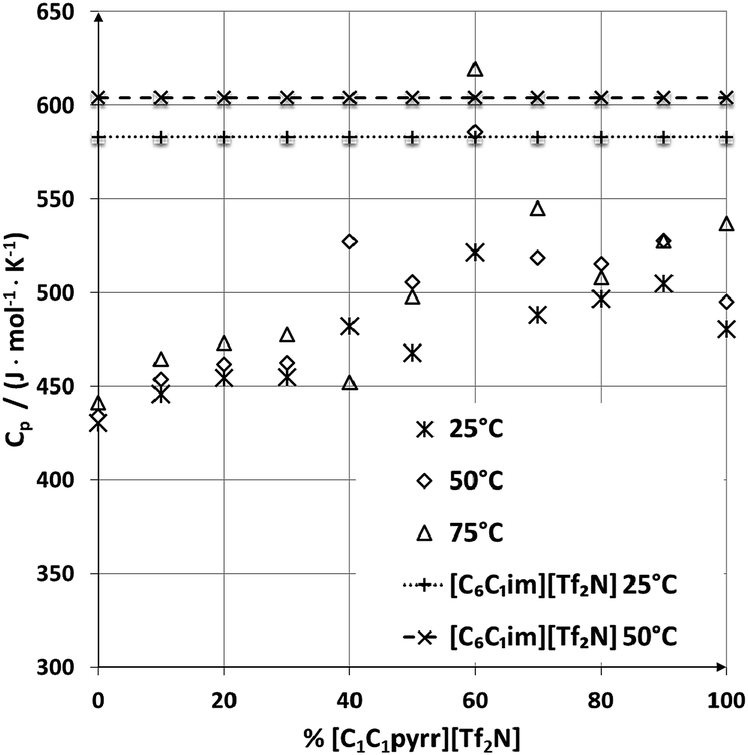 | ||
| Fig. 4 The heat capacities for all samples measured at 25, 50, and 75 °C. Data for [C6C1im][Tf2N] at 25 and 50 °C is shown for comparison.18 Error bars are omitted for clarity (error = 1%). | ||
Conductivity
The electrical conductivity of IL compounds determines their usefulness in electrochemical applications. The three binary combinations (10%–30% [C1C1pyrr][Tf2N]) that were stable liquids at 50 °C were measured for conductivity at this temperature. The results ranged from 11.2 to 12.2 mS cm−1, which is a three-fold improvement over [C6C1im][Tf2N]'s conductivity of 4.1 mS cm−1 (Table 1). This could be partially due to the increased charge per unit volume that results from smaller cations, but the significant boost suggests other mechanisms are at work as well. An alternate explanation for the increase in conductivity may be the inability of this system to form any ionic liquid aggregates and nano-domains25,35 due to the lack of hydrocarbon chains. Their high conductivity makes these binary mixtures potential candidates for electrochemical applications.Two properties of these novel ionic liquid mixtures are particularly interesting. First, the conductivity is nearly three times that of [C6C1im][Tf2N] even though the dynamic viscosity is the same within 5%. Second, the solubility of CO2 is comparable between the mixtures and [C6C1im][Tf2N], despite a 15% increase in density.
Molecular insights via 2D-IR spectroscopy
To gain a better understanding of the microscopic causes of these macroscopic observations, we used 2D-IR spectroscopy to probe the picosecond dynamics of the mixtures compared with [C6C1im][Tf2N]. 2D-IR spectroscopy is a coherent vibrational spectroscopy, which is analogous to 2D NMR spectroscopy. Where 2D NMR correlates nuclear spins, 2D-IR correlates molecular vibrational frequencies. The 2D-IR line shape encodes the rate at which the local environment changes around a molecule.Not all molecular vibrations are sufficiently intense to observe in a 2D-IR spectrum; CO2, however, is an ideal vibrational chromophore for 2D-IR in ionic liquids.27 The vibrational spectrum of the CO2 asymmetric stretch depends on local intermolecular interactions such as electrostatic, charge transfer, and hydrogen bonding.
We used the CO2 asymmetric stretch as a probe for 2D-IR spectroscopy to investigate the differences in the local environment experienced by CO2 in both [C1C1pyrr]0.3[C1pyr]0.7[Tf2N] and [C6C1im][Tf2N] (Fig. 5). While this experiment has direct motivation from the potential use of these novel ionic liquids for carbon capture, this molecular probe also allowed us to investigate the local motions of the cation and anion, which are important for understanding the differences in conductivity between the ILs, and which CO2 does not greatly disrupt. The microscopic dynamics in the two ILs are markedly different. Spectra of the CO2 asymmetric stretch in both ILs start to elongate in the diagonal direction (Fig. 5A and D), which indicates a distribution of local environments. At 5 ps, the 2D-IR spectrum of [C1C1pyrr]0.3[C1pyr]0.7[Tf2N] becomes round, indicating that the environment around the CO2 molecule was randomized (Fig. 5B), whereas the 2D-IR spectrum of [C6C1im][Tf2N] retains some diagonal character (Fig. 5E), indicating that the environment around the CO2 molecules remains similar. In [C6C1im][Tf2N], the spectrum becomes round at a 50 ps time scale (Fig. 5F).
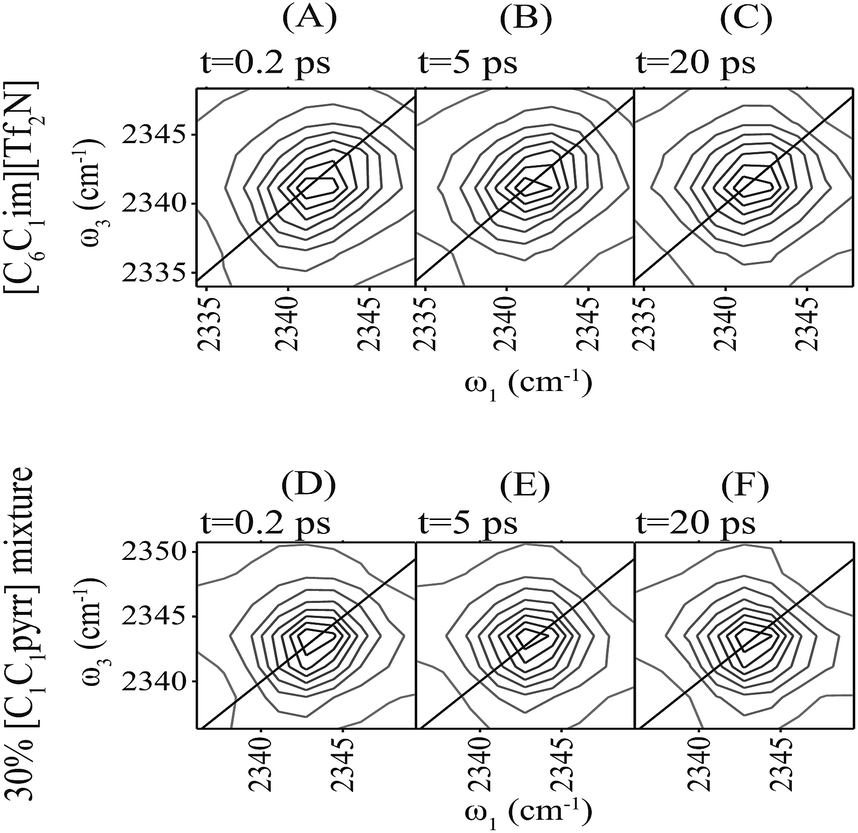 | ||
| Fig. 5 2D-IR spectra of the CO2 anti-symmetric stretch in [C6C1im][Tf2N] and in [C1C1pyrr]0.3[C1pyr]0.7[Tf2N]. | ||
This qualitative picture can be quantified by non-linear least squares fitting of the spectra to third-order response functions.36 The three important parameters from the analysis are the dephasing time (T2), which controls the anti-diagonal width of the spectra; the inhomogeneous width (Δ), which controls the diagonal width of the spectra; and the spectral diffusion time (τc), which controls the rate of change of shape of the spectra. T2 is related to fast local motions such as hindered rotations (librations), Δ is related to the distribution of different environments, and τc is the time scale for relaxation of those initial environments. In [C1C1pyrr]0.3[C1pyr]0.7[Tf2N], CO2 shows a marked decrease in τc and an increase in T2 compared with that seen in [C6C1im][Tf2N]. The difference in τc we attributed to differences in the rotational ease of the ions of CO2's first solvent shell. The increased dephasing time, T2, likely results from a narrower distribution of CO2 frequencies during homogeneous motions, perhaps due to the loss of largely uncharged hexyl chains in the first solvation shell of [C1C1pyrr]0.3[C1pyr]0.7[Tf2N]. Δ is similar for the two ILs (Table 2).
| Sample | Δ (cm−1) | τc (ps) | T2 (ps) |
|---|---|---|---|
| [C6C1im][Tf2N] | 1.4 ± 0.1 | 47 ± 9 | 2.45 ± 0.03 |
| [C1C1pyrr]0.3[C1pyr]0.7[Tf2N] | 1.5 ± 0.1 | 6 ± 2 | 3.0 ± 0.2 |
The most striking spectroscopic difference is the eight-fold decrease in τc between CO2 in [C6C1im][Tf2N] and [C1pyr]0.7[C1C1pyrr]0.3[Tf2N]. Previous work has identified that, in a range of imidazolium ionic liquids, τc for CO2 is correlated to the solvent viscosity.36 CO2 in [C6C1im][Tf2N], as expected, falls on this trend line. The IL mixture, however, despite a similar viscosity, shows considerably faster relaxation of the local structure around CO2, as evidenced by τc. We hypothesize that the removal of hexyl chains in the novel mixtures removes the steric hindrance to rotational motions of the cation. Therefore, CO2 can move relatively quickly between different local environments.
This spectroscopic finding also provides insight into a possible molecular mechanism for the increased conductivity seen in [C1pyr]0.7[C1C1pyrr]0.3[Tf2N] compared to [C6C1im][Tf2N]. Under an external potential, the decreased energy barrier to solvent reorientation, as described above, should allow easier molecular reorientation to permit ion transport and thus conduction. This effect, combined with decreased intermolecular friction due to the lack of bulky alkyl chains, and increased charge density (due to decreased IL molar volume), provide a plausible explanation for the increased conductivity. It may be possible to exploit combinations of these mechanisms to guide the design of ionic liquids with high conductivity for electrochemical applications.
Conclusions
By mixing two dissimilar cations with shortened alkyl chains, useful properties can be accessed and tuned. Mixing the two compounds increases the mobility of the cation, which leads to greatly increased conductivity that is promising for electrochemical applications. The removal of the hexyl chain also decreases the heat capacity of the IL, which can be important for variety of thermal applications. Despite an increase in density, the CO2 solubility of the novel compounds and mixtures remains comparable to that of [C6C1im][Tf2N] and the best mixture depresses the melting point below 35 °C. The melting point and CO2 solubility can likely be improved with further experimentation. This approach shows promise as a general strategy to inexpensively optimize the properties of ionic liquids by mixing simple salts, rather than the complicated synthesis of new ILs.Acknowledgements
This research was supported by the U.S. Department of Energy's National Energy Technology Laboratory under the contract DE-FE0004000. Part of this work was also supported by ACS PRF Award #53936-DNI6.Notes and references
- P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772–3789 CrossRef CAS.
- J. E. Brennecke and B. E. Gurkan, J. Phys. Chem. Lett., 2010, 1, 3459–3464 CrossRef CAS.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS PubMed.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- H. B. Nulwala, C. N. Tang, B. W. Kail, K. Damodaran, P. Kaur, S. Wickramanayake, W. Shi and D. R. Luebke, Green Chem., 2011, 13, 3345–3349 RSC.
- C. Wang, X. Luo, H. Luo, D. Jiang, H. Li and S. Dai, Angew. Chem., Int. Ed., 2011, 50, 4918–4922 CrossRef CAS PubMed.
- G. Cui, J. Zheng, X. Luo, W. Lin, F. Ding, H. Li and C. Wang, Angew. Chem., 2013, 125, 10814–10818 CrossRef PubMed.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS PubMed.
- K. Fukumoto, M. Yoshizawa and H. Ohno, J. Am. Chem. Soc., 2005, 127, 2398–2399 CrossRef CAS PubMed.
- B. E. Gurkan, J. C. de la Fuente, E. M. Mindrup, L. E. Ficke, B. F. Goodrich, E. a. Price, W. F. Schneider and J. F. Brennecke, J. Am. Chem. Soc., 2010, 132, 2116–2117 CrossRef CAS PubMed.
- W. Ogihara, M. Yoshizawa and H. Ohno, Chem. Lett., 2004, 33, 1022–1023 CrossRef CAS.
- X. Luo, Y. Guo, F. Ding, H. Zhao, G. Cui, H. Li and C. Wang, Angew. Chem., Int. Ed., 2014, 53, 7053–7057 CrossRef CAS PubMed.
- K. A. Fletcher, S. N. Baker, G. A. Baker and S. Pandey, New J. Chem., 2003, 27, 1706–1712 RSC.
- P. M. Bayley, A. S. Best, D. R. MacFarlane and M. Forsyth, ChemPhysChem, 2011, 12, 823–827 CrossRef CAS PubMed.
- H. J. Castejón and R. J. Lashock, J. Mol. Liq., 2012, 167, 1–4 CrossRef PubMed.
- H. Niedermeyer, J. P. Hallett, I. J. Villar-Garcia, P. A. Hunt and T. Welton, Chem. Soc. Rev., 2012, 41, 7780–7802 RSC.
- G. Chatel, J. F. B. Pereira, V. Debbeti, H. Wang and R. D. Rogers, Green Chem., 2014, 16, 2051–2083 RSC.
- J. M. Crosthwaite, M. J. Muldoon, J. K. Dixon, J. L. Anderson and J. F. Brennecke, J. Chem. Thermodyn., 2005, 37, 559–568 CrossRef CAS PubMed.
- J. N. C. Lopes, A. A. H. Padua and K. Shimizu, J. Phys. Chem. B, 2008, 112, 5039–5046 CrossRef PubMed.
- J. L. Anderson, J. K. Dixon and J. F. Brennecke, Acc. Chem. Res., 2007, 40, 1208–1216 CrossRef CAS PubMed.
- J. A. Widegren and J. W. Magee, J. Chem. Eng. Data, 2007, 52, 2331–2338 CrossRef CAS.
- C. Schreiner, S. Zugmann, R. Hartl and H. J. Gores, J. Chem. Eng. Data, 2010, 49, 4372–4377 CrossRef.
- G. J. Maximo, R. J. B. N. Santos, P. Brandão, J. M. S. S. Esperança, M. C. Costa, A. J. A. Meirelles, M. G. Freire and J. A. P. Coutinho, Cryst. Growth Des., 2014, 14, 4270–4277 CAS.
- Y. Chen, F. Ke, H. Wang, Y. Zhang and D. Liang, ChemPhysChem, 2012, 13, 160–167 CrossRef CAS PubMed.
- J. N. Canongia Lopes and A. A. H. Padua, J. Phys. Chem. B, 2006, 110, 19586–19592 CrossRef CAS PubMed.
- Y. Wang and G. A. Voth, J. Am. Chem. Soc., 2005, 127, 12192–12193 CrossRef CAS PubMed.
- A. M. Pinto, H. Rodríguez, Y. J. Colón, A. Arce and A. Soto, Ind. Eng. Chem. Res., 2013, 52, 5975–5984 CrossRef CAS.
- Y.-F. Hu, Z.-C. Liu, C.-M. Xu and X.-M. Zhang, Chem. Soc. Rev., 2011, 40, 3802–3823 RSC.
- J. M. Pringle, P. C. Howlett, D. R. MacFarlane and M. Forsyth, J. Mater. Chem., 2010, 20, 2056 RSC.
- C. M. Gordon, J. D. Holbrey, A. R. Kennedy and K. R. Seddon, J. Mater. Chem., 1998, 8, 2627–2636 RSC.
- R. D. Chirico, V. Diky, J. W. Magee, M. Frenkel and K. N. Marsh, Pure Appl. Chem., 2009, 81, 791–828 CrossRef CAS.
- K. N. Marsh, J. F. Brennecke, R. D. Chirico, M. Frenkel, A. Heintz, J. W. Magee, C. J. Peters, L. P. N. Rebelo and K. R. Seddon, Pure Appl. Chem., 2009, 81, 781–790 CrossRef CAS.
- M. J. Muldoon, S. Aki, J. L. Anderson, J. K. Dixon and J. F. Brennecke, J. Phys. Chem. B, 2007, 111, 9001–9009 CrossRef CAS PubMed.
- M. B. Shiflett and A. Yokozeki, J. Phys. Chem. B, 2007, 111, 2070–2074 CrossRef CAS PubMed.
- A. Seduraman, M. Klähn and P. Wu, Calphad, 2009, 33, 605–613 CrossRef CAS PubMed.
- T. Brinzer, E. J. Berquist, Z. Ren, S. Dutta, C. A. Johnson, C. S. Krisher, D. S. Lambrecht and S. Garrett-Roe, J. Chem. Phys., 2015, 142, 212425 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra06561e |
| This journal is © The Royal Society of Chemistry 2015 |