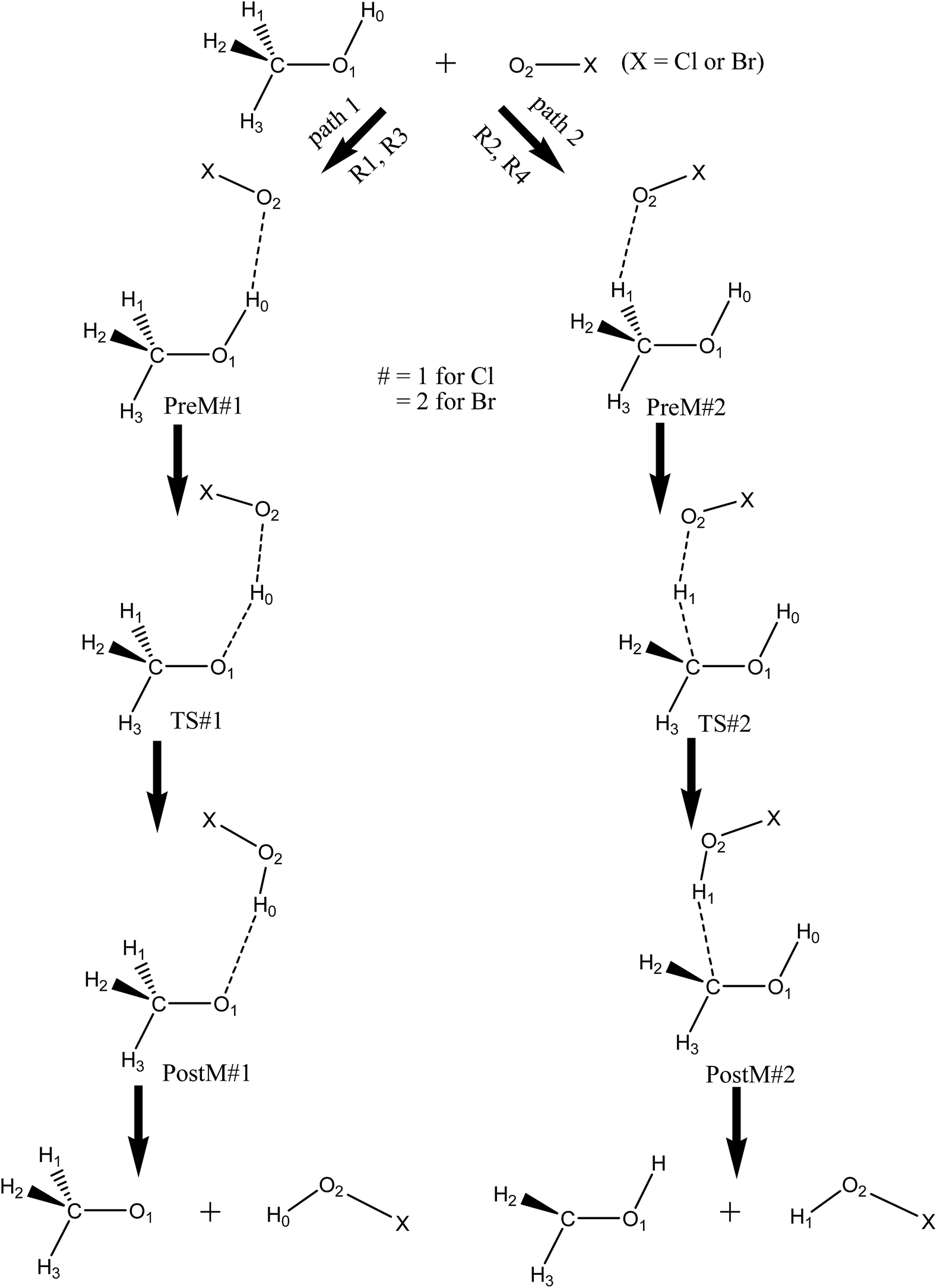
DOI:
10.1039/C5RA06483J
(Paper)
RSC Adv., 2015,
5, 39110-39121
Theoretical studies on gas-phase kinetics and mechanism of H-abstraction reaction from methanol by ClO and BrO radicals†
Received
15th February 2015
, Accepted 22nd April 2015
First published on 22nd April 2015
Abstract
The gas-phase kinetics and mechanism of two channel hydrogen (H) abstraction reaction of methanol (CH3OH) by halogen monoxide (XO, X = Cl, Br) radical has been investigated using theoretical approach. The two H-abstraction channels followed are: hydroxyl H-atom or methyl H-atom of methanol (CH3OH). The geometry optimization and frequency calculations were performed at M06-2X method and cc-pVTZ basis set. Single point energy calculation of all the species were computed at high level CCSD(T)/cc-pVTZ theory on the M06-2X/cc-pVTZ optimized structure. Weak intermolecular pre-reactive and post-reactive complexes were located at the entrance and exit channel respectively on the potential energy surfaces of all the H-abstraction reactions. The rate constants (k) and branching ratio (ϕ) of all possible channels were calculated as a function of temperature for a wide range of temperature 200–2500 K. The rate constants were estimated using canonical variational transition state theory (CVT) combined with an Eckart tunneling correction and hindered rotor approximation for low frequency torsional modes. The rate constant calculation shows that the reaction for the H-abstraction from methyl group of CH3OH by XO radical leading to hydroxymethyl (CH2OH) radical is predominant over the hydroxyl group H-abstraction reaction forming methoxy (CH3O) radical. Arrhenius equation using three parameters was obtained by fitting the kinetic data for the two channels of H-abstraction by ClO and BrO radicals. The overall rate expression, in units of cm3 per molecule per s, with ClO radical is found to be kov1(T) = 3.92 × 10−19T1.63![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−2062/T) and with BrO radical it is kov2(T) = 1.53 × 10−20T2.41exp(−2206/T) for the temperature range 200–2500 K.
exp(−2062/T) and with BrO radical it is kov2(T) = 1.53 × 10−20T2.41exp(−2206/T) for the temperature range 200–2500 K.
1. Introduction
Methanol (CH3OH) is an oxygenated volatile liquid mostly used as common organic solvent in chemical laboratories and as fuel directly or as a fuel additive with gasoline. It is considered as an alternate fuel as it produces lesser air pollutants in combustion than gasoline.1,2 Methanol is the most abundant non-methane oxygenated volatile gas in the atmosphere with the estimated global emissions3 up to 240 Tg per year. In atmosphere, the methanol emission sources includes the plant growth, biomass burning, biofuels, industry, vehicles, plant decay and atmospheric reactions.3–5 The major sink of methanol released into the atmosphere is oxidation by the hydroxyl (OH) radical with atmospheric lifetime of about 10 days.3 This comparatively longer lifetime may cause it to interact with other atmospheric species. Due to its large emission, it is important to know the fate of methanol in atmosphere.
The chlorinated or brominated species are important from both atmospheric and combustion chemistry point of view. In atmosphere halogen oxide radicals, XO (X = Cl or Br) are formed from the reaction of halogen atom with ozone, heterogeneous oxidation of halogen atom or atmospheric degradation of X containing species (for example, formation of ClO from CFCs, CH3Cl).6–8 Chlorine monoxide (ClO) is also detected at the same level of OH radical concentration near the surface of the burner in ammonium perchlorate combustion process in a hydrogen atmosphere at 1400 K.9,10 The O2–H2 flame doped with X2 shows the presence of XO radicals.11,12 There is no information regarding kinetics and mechanism of CH3OH + XO reaction. The increasing use of CH3OH as direct fuel or as a fuel additive enhances the presence of it in the atmosphere, thereby augments the significance of this study.
The presence of two nonequivalent hydrogen (H) atoms in CH3OH leads to the formation of two structural isomers, methoxy (CH3O) or hydroxymethyl (CH2OH), which are the product radicals. These product radicals are produced by competitive hydroxyl H-abstraction or methyl H-abstraction reaction by XO radicals. In this study, the two possible H-abstraction reaction channels from CH3OH by XO radical to produce either CH3O or CH2OH radicals have been studied. The possible pathways are as follows:
| | | CH3OH + ClO → CH3O + HOCl | (R1) |
| | | CH3OH + ClO → CH2OH + HOCl | (R2) |
| | | CH3OH + BrO → CH3O + HOBr | (R3) |
and
| | | CH3OH + BrO → CH2OH + HOBr | (R4) |
The present study focuses on comprehensive insight of two possible H-abstraction reactions, R1 to R4 of CH3OH with ClO and BrO radical using theoretical approach. The different thermodynamic and kinetic parameters of the individual pathways are also discussed over a wide temperature range and is required for atmospheric and combustion reaction modeling. The relative formations of the methoxy and hydroxymethyl radicals are emphasized by the branching ratios. The temperature dependence of branching ratios is important to understand the contribution of the two competitive pathways towards the overall rate constants. Theoretical studies of kinetics and mechanism of the reaction of CH3OH with ClO and BrO radicals is a necessary step which will aid experimentalist over a wide range of temperature.
2. Computational methods
Geometry optimization of the reactants (CH3OH, XO (X = Cl, Br)), pre- and post-reactive molecular complexes (PreM and PostM respectively), transition states (TS), and product molecules (CH3O, CH2OH, HOX) involved in the H-abstraction reaction from CH3OH by XO radical were carried out employing the Minnesota hybrid meta density functional theory (DFT) level of M06-2X,13,14 and Dunning's correlation-consistent polarized valence triple-zeta, cc-pVTZ basis set. M06-2X functional is found to be good performer for main group chemistry, in predicting transition state geometry calculation and also performs well in calculating non-covalent interactions15 and thermochemistry and kinetic parameters.13,14,16 Frequency calculations of all the species were executed at the same level of theory to judge the stationary points on the potential energy surface as minima (NIMAG = 0) or transition states (NIMAG = 1). In case of the transition states, the corresponding bond breaking and forming is reflected by the presence of one imaginary frequency associated with it. Intrinsic reaction coordinate (IRC)17,18 analyses were performed at the same level of theory to obtain the minimum energy path (MEP) of the reactions and to ascertain whether the transition states connect different local minima along the reaction path. Based on the M06-2X/cc-pVTZ optimized structures, the potential energy surface of the titled reaction was refined with single point energy calculation at high level coupled cluster method with single and double excitations and perturbative triples, CCSD(T)/cc-pVTZ. The zero-point energies (ZPE) obtained at M06-2X/cc-pVTZ was added to CCSD(T)/cc-pVTZ energies to obtain the different ZPE corrected energies. All reported calculations were performed employing Gaussian 09 package of programs19 and GaussView 5.0.9 software20 was used for the visualization of molecular structures, orbitals and vibrational motions. The spin expectation value, 〈S2〉, at M06-2X level for all doublet systems remained in the range of 0.75–1.00. So, spin contamination was negligible during the progress of the reaction via four different reaction paths R1, R2, R3 and R4. PreMs were found at the entrance channel of the reaction and PostMs were at the exit channel of the reaction. The PreM and PostM complexes have all real frequencies.
The reaction rates were evaluated using canonical variational transition state theory (CVT)21 with the inclusion of unsymmetrical one dimensional tunneling correction by Eckart22 over the temperature range 200–2500 K. The rate constants were computed using KISTHELP23 program. The CVT rate constant, kCVT(T) is estimated by minimizing generalized transition state24 rate constant, kGT(T,s) with respect to reaction coordinate s. s represents the distance along the MEP from the saddle point. Thus, kCVT(T) with the Eckart tunneling correction χ(T) is given by
here,
kb is the Boltzmann's constant,
T is the temperature,
h is the Planck's constant,
NA is Avogadro's number,
V# is potential energy barrier including ZPE correction.
σ is the symmetry factor which denotes the reaction path degeneracy.
σ is 1 and 3 respectively for H-abstraction from hydroxyl group (path 1) and methyl group (path 2) of CH
3OH.
QTS and
QR are total partition function of the TS and reactants per unit volume. The total partition function terms are consisted of translational, electronic, rotational and vibrational partition functions. Rigid-rotor harmonic oscillator approximation has been used. The low frequency torsional modes of the reactants and the transition states have been treated using hindered rotor approximation while calculating their contribution to the partition functions. Relaxed potential energy scan at M06-2X/cc-pVTZ using the optimized structures at same level have been performed for the low frequency torsional modes to calculate corresponding rotational barrier height of internal rotation. As in our previous study,
25 the rate constant expression does not contain the energy of the molecular complexes and the same argument is applied here.
Spin–orbit effect is important in case of halogen atoms.26,27 The spin–orbit correction of −0.92 kcal per mol for ClO28 and −2.79 kcal per mol for BrO29 were taken into account. The potential energies of ClO radical and pre-reactive molecular complexes (PreM11, PreM12) involving ClO are associated with the spin–orbit correction of −0.92 kcal per mol while BrO and BrO related pre-reactive molecular complexes (PreM21, PreM22) are having −2.79 kcal per mol spin–orbit correction value.
The bonding features of different stationary points on potential energy surface of CH3OH + XO (X = Cl, Br) reaction were studied in terms of electronic density at M06-2X/cc-pVTZ using atoms in molecules (AIM) approach by Bader30,31 as implemented in AIMALL suite of program.32 The wave function required for AIM calculation was generated from Gaussian 09.
3. Results and discussions
3.1. Geometries and vibrational frequencies of the stationary points
The detailed Cartesian coordinates of the optimized reactants, pre-reactive (PreM) and post-reactive (PostM) molecular complexes, transition states (TS) and product radicals at M06-2X/cc-pVTZ level of theory are given in ESI Table 1† and additional structural parameters in ESI Fig. 1.† The schematic representation of the two H-abstraction reaction pathways from CH3OH by XO radical is shown in Fig. 1 and related structural parameters at M06-2X/cc-pVTZ are reported in Table 1. The potential energy surface of the reactions R1 to R4 with zero point energy correction at CCSD(T)/cc-pVTZ//MO6-2X/cc-pVTZ is shown in Fig. 2 and 3. Table 2 shows calculated rotational constants and the vibrational frequencies of different stationary points at M06-2X/cc-pVTZ. The electron density, ρ at bond critical points (BCP) of the chemical bonds (intra or inter) present in all corresponding stationary points of CH3OH + XO (X = Cl, Br) reaction is summarized in Table 3.
 |
| | Fig. 1 Schematic representation of the CH3OH + XO (X = Cl, Br) reaction. | |
Table 1 Structural parameters (r1, r2 are in Å and a1, θ, ϕ are in degrees) of the molecular complexes and transition states for the two different pathways for the reaction of CH3OH with XO (X = Cl or Br) radical computed at M06-2X/cc-pVTZ
| Parameters |
CH3OH + ClO |
CH3OH + BrO |
| PreM11 |
TS11 |
PostM11 |
PreM12 |
TS12 |
PostM12 |
PreM21 |
TS21 |
PostM21 |
PreM22 |
TS22 |
PostM22 |
|
r(Cl–O2) |
1.567 |
1.632 |
1.677 |
1.567 |
1.637 |
1.675 |
|
|
|
|
|
|
|
r(Br–O2) |
|
|
|
|
|
|
1.709 |
1.761 |
1.811 |
1.708 |
1.770 |
1.809 |
|
r(C–O1) |
1.410 |
1.379 |
1.372 |
1.415 |
1.371 |
1.352 |
1.410 |
1.372 |
1.372 |
1.416 |
1.374 |
1.353 |
|
r(C–H1) |
1.095 |
1.102 |
1.093 |
1.093 |
1.232 |
2.131 |
1.095 |
1.110 |
1.093 |
1.093 |
1.224 |
2.150 |
|
r(C–H2) |
1.094 |
1.092 |
1.103 |
1.093 |
1.092 |
1.083 |
1.093 |
1.094 |
1.103 |
1.093 |
1.092 |
1.083 |
|
r(C–H3) |
1.088 |
1.098 |
1.093 |
1.088 |
1.086 |
1.079 |
1.088 |
1.094 |
1.093 |
1.088 |
1.086 |
1.079 |
|
r(O1–H0) |
0.961 |
1.185 |
1.845 |
0.959 |
0.962 |
0.964 |
0.961 |
1.188 |
1.862 |
0.959 |
0.962 |
0.964 |
|
r(H0–O2) |
2.157 |
1.147 |
0.976 |
|
|
|
2.115 |
1.156 |
0.975 |
|
|
|
|
r(H1–O2) |
|
|
|
2.593 |
1.328 |
0.977 |
|
|
|
2.569 |
1.341 |
0.976 |
|
r(Cl–H0) |
|
|
|
|
|
2.607 |
|
|
|
|
|
|
|
r(Br–H0) |
|
|
|
|
|
|
|
|
|
|
|
2.751 |
|
r(Cl–H1) |
|
2.645 |
2.951 |
|
|
|
|
|
|
|
|
|
|
r(Br–H1) |
|
|
|
|
|
|
|
2.630 |
|
|
|
|
|
r(Cl–O1) |
|
|
|
3.112 |
3.149 |
|
|
|
|
|
|
|
|
r(Br–O1) |
|
|
|
|
|
|
|
|
|
3.217 |
3.253 |
|
|
θ(C–O1–H0) |
107.8 |
112.4 |
104.1 |
108.6 |
109.9 |
109.7 |
107.7 |
111.8 |
103.7 |
108.6 |
109.9 |
109.7 |
|
θ(O1–C–H1) |
112.1 |
110.7 |
112.9 |
112.1 |
108.9 |
92.5 |
112.1 |
109.5 |
112.9 |
112.1 |
109.3 |
92.9 |
|
θ(O1–C–H2) |
112.2 |
113.2 |
105.0 |
111.9 |
115.0 |
118.5 |
112.2 |
109.7 |
105.0 |
111.8 |
114.8 |
118.5 |
|
θ(O1–C–H3) |
107.4 |
107.3 |
111.5 |
107.0 |
109.9 |
113.6 |
107.5 |
113.2 |
111.5 |
107.1 |
109.6 |
113.6 |
|
θ(O1–H0–O2) |
141.0 |
165.6 |
157.2 |
|
|
|
143.8 |
164.7 |
156.6 |
|
|
|
|
θ(H0–O2–Cl) |
97.7 |
107.6 |
102.5 |
|
|
|
|
|
|
|
|
|
|
θ(H0–O2–Br) |
|
|
|
|
|
|
99.1 |
110.1 |
102.7 |
|
|
|
|
θ(O2–H1–C) |
|
|
|
107.9 |
166.4 |
152.0 |
|
|
|
107.9 |
168.3 |
153.5 |
|
θ(Cl–O2–H1) |
|
|
|
99.4 |
104.6 |
103.4 |
|
|
|
|
|
|
|
θ(Br–O2–H1) |
|
|
|
|
|
|
|
|
|
103.0 |
105.6 |
103.8 |
|
ϕ(H0–O1–C–H1) |
58.7 |
38.1 |
19.1 |
−59.9 |
−70.2 |
−63.6 |
−57.6 |
39.3 |
−19.0 |
−59.5 |
−71.3 |
−65.6 |
|
ϕ(H0–O1–C–H2) |
−63.7 |
−86.3 |
−98.3 |
63.0 |
45.2 |
27.6 |
64.7 |
154.6 |
98.3 |
63.5 |
44.9 |
27.2 |
|
ϕ(H0–O1–C–H3) |
177.4 |
152.2 |
146.8 |
−178.2 |
174.6 |
177.4 |
−176.3 |
−80.4 |
−146.7 |
−177.7 |
173.6 |
177.1 |
|
ϕ(O2–H0–O1–C) |
−64.3 |
2.9 |
−28.0 |
|
|
|
66.1 |
−73.5 |
30.0 |
|
|
|
|
ϕ(Cl–O2–H0–O1) |
−32.0 |
−57.3 |
−51.5 |
|
|
|
|
|
|
|
|
|
|
ϕ(Br–O2–H0–O1) |
|
|
|
|
|
|
30.6 |
68.6 |
54.3 |
|
|
|
|
ϕ(O2–H1–C–O1) |
|
|
|
−75.7 |
37.0 |
53.8 |
|
|
|
−76.4 |
32.2 |
53.3 |
|
ϕ(Cl–O2–H1–C) |
|
|
|
50.2 |
−15.8 |
12.6 |
|
|
|
|
|
|
|
ϕ(Br–O2–H1–C) |
|
|
|
|
|
|
|
|
|
52.1 |
−15.5 |
15.2 |
|
L
|
|
1.2 |
|
|
0.4 |
|
|
1.2 |
|
|
0.3 |
|
 |
| | Fig. 2 Potential energy surface of CH3OH + ClO reaction (R1, R2) at CCSD(T)/cc-pVTZ//M06-2X/cc-pVTZ. | |
 |
| | Fig. 3 Potential energy surface of CH3OH + BrO reaction (R3, R4) at CCSD(T)/cc-pVTZ//M06-2X/cc-pVTZ. | |
Table 2 Calculated rotational constants (MHz) and unscaled vibrational frequencies (cm−1) at M06-2X/cc-pVTZ level for the reactants, transition states, molecular complexes and products
| Species |
Rotational constants (in MHz) |
Vibrational frequencies (in cm−1) |
| CH3OH |
129091, 24995, 24126 |
255, 1071, 1119, 1183, 1374, 1489, 1509, 1520, 3024, 3078, 3145, 3910 |
| OCl |
18671 |
900 |
| OBr |
12939 |
784 |
| PreM11 |
11809, 2222, 2123 |
61, 75, 82, 163, 170, 534, 924, 1096, 1125, 1185, 1417, 1491, 1513, 1525, 3020, 3071, 3138, 3886 |
| PreM12 |
10608, 2573, 2125 |
40, 48, 79, 103, 141, 306, 914, 1070, 1110, 1184, 1373, 1490, 1503, 1522, 3034, 3090, 3145, 3900 |
| PreM21 |
10147, 1672, 1577 |
44, 66, 73, 111, 176, 484, 785, 1084, 1121, 1185, 1392, 1489, 1507, 1522, 3023, 3073, 3138, 3863 |
| PreM22 |
8909, 1954, 1635 |
45, 48, 91, 98, 133, 323, 784, 1071, 1109, 1183, 1374, 1491, 1502, 1518, 3035, 3092, 3146, 3899 |
| TS11 |
12369, 2850, 2459 |
1946i, 60, 118, 157, 272, 532, 784, 865, 1063, 1110, 1191, 1356, 1404, 1449, 1496, 2957, 3023, 3098 |
| TS12 |
10914, 2935, 2375 |
1250i, 35, 133, 276, 348, 499, 634, 882, 1092, 1178, 1232, 1323, 1390, 1450, 1504, 3061, 3170, 3860 |
| TS21 |
10560, 2117, 1785 |
1918i, 64, 88, 160, 307, 510, 737, 894, 1057, 1124, 1171, 1386, 1423, 1473, 1504, 2870, 3032, 3091 |
| TS22 |
9531, 2115, 1762 |
1124i, 88, 118, 275, 335, 497, 630, 830, 1085, 1179, 1239, 1328, 1386, 1442, 1505, 3064, 3177, 3865 |
| postM11 |
11376, 2376, 2181 |
29, 68, 96, 123, 229, 603, 806, 927, 1018, 1131, 1372, 1385, 1408, 1519, 2956, 3049, 3105, 3610 |
| PostM12 |
10350, 2571, 2287 |
64, 124, 134, 182, 245, 408, 565, 757, 804, 1084, 1257, 1360, 1380, 1497, 3139, 3279, 3564, 3840 |
| PostM21 |
9900, 1740, 1609 |
41, 70, 96, 125, 224, 593, 701, 926, 1017, 1131, 1323, 1377, 1405, 1518, 2958, 3049, 3103, 3635 |
| PostM22 |
8735, 1911, 1705 |
57, 104, 126, 178, 238, 403, 557, 700, 754, 1081, 1255, 1294, 1374, 1496, 3140, 3280, 3592, 3836 |
| CH3O |
158442, 28006, 27835 |
237, 972, 1137, 1384, 1389, 1526, 2966, 3036, 3077 |
| CH2OH |
194266, 30120, 26338 |
412, 596, 1066, 1240, 1365, 1496, 3143, 3280, 3906 |
| HOCl |
619444, 15350, 14979 |
822, 1284, 3856 |
| HOBr |
620433, 10758, 10574 |
692, 1212, 3860 |
Table 3 The AIM calculated electron density (ρ, in au) of bond critical points (BCP) of the stationary points of CH3OH + XO (X = Cl, Br) reaction computed at the level of M06-2X/cc-pVTZ
| CH3OH + ClO reaction |
| Parameters |
Reactants |
Products |
R1
|
R2
|
| CH3OH |
ClO |
CH3O |
CH2OH |
HOCl |
PreM11 |
TS11 |
PostM11 |
PreM12 |
TS12 |
PostM12 |
|
ρ(Cl–O2) |
|
0.278 |
|
|
0.218 |
0.280 |
0.243 |
0.220 |
0.280 |
0.240 |
0.222 |
|
ρ(O2–H) |
|
|
|
|
0.367 |
|
|
|
|
|
|
|
ρ(C–O1) |
0.260 |
|
0.290 |
0.282 |
|
0.262 |
0.277 |
0.283 |
0.257 |
0.279 |
0.289 |
|
ρ(C–H1) |
0.289 |
|
0.287 |
|
|
0.287 |
0.280 |
0.291 |
0.289 |
0.196 |
0.022 |
|
ρ(C–H2) |
0.289 |
|
0.287 |
0.294 |
|
0.289 |
0.292 |
0.273 |
0.290 |
0.290 |
0.293 |
|
ρ(C–H3) |
0.293 |
|
0.274 |
0.298 |
|
0.293 |
0.281 |
0.288 |
0.293 |
0.295 |
0.298 |
|
ρ(O1–H0) |
0.372 |
|
|
0.371 |
|
0.369 |
0.176 |
0.033 |
0.372 |
0.367 |
0.364 |
|
ρ(H0–O2) |
|
|
|
|
|
0.016 |
0.202 |
0.350 |
|
|
|
|
ρ(H1–O2) |
|
|
|
|
|
|
|
|
0.008 |
0.131 |
0.349 |
|
ρ(Cl–H0) |
|
|
|
|
|
|
|
|
|
|
0.012 |
|
ρ(Cl–O1) |
|
|
|
|
|
|
|
|
0.010 |
0.010 |
|
|
ρ(Cl–H1) |
|
|
|
|
|
|
0.011 |
0.006 |
|
|
|
| CH3OH + BrO reaction |
| Parameters |
Reactants |
Products |
R3
|
R4
|
| CH3OH |
BrO |
CH3O |
CH2OH |
HOBr |
PreM21 |
TS21 |
PostM21 |
PreM22 |
TS22 |
PostM22 |
|
ρ(Br–O2) |
|
0.211 |
|
|
0.170 |
0.212 |
0.191 |
0.173 |
0.213 |
0.188 |
0.174 |
|
ρ(O2–H) |
|
|
|
|
0.366 |
|
|
|
|
|
|
|
ρ(C–O1) |
0.260 |
|
0.290 |
0.282 |
|
0.262 |
0.282 |
0.283 |
0.257 |
0.278 |
0.288 |
|
ρ(C–H1) |
0.289 |
|
0.287 |
|
|
0.287 |
0.270 |
0.290 |
0.290 |
0.201 |
0.021 |
|
ρ(C–H2) |
0.289 |
|
0.287 |
0.294 |
|
0.289 |
0.287 |
0.273 |
0.289 |
0.290 |
0.293 |
|
ρ(C–H3) |
0.293 |
|
0.274 |
0.298 |
|
0.293 |
0.290 |
0.288 |
0.294 |
0.295 |
0.298 |
|
ρ(O1–H0) |
0.372 |
|
|
0.371 |
|
0.368 |
0.176 |
0.032 |
0.372 |
0.367 |
0.364 |
|
ρ(H0–O2) |
|
|
|
|
|
0.018 |
0.196 |
0.351 |
|
|
|
|
ρ(H1–O2) |
|
|
|
|
|
|
|
|
0.008 |
0.126 |
0.350 |
|
ρ(Br–H0) |
|
|
|
|
|
|
|
|
|
|
0.011 |
|
ρ(Br–O1) |
|
|
|
|
|
|
|
|
0.010 |
0.010 |
|
|
ρ(Br–H1) |
|
|
|
|
|
|
0.015 |
|
|
|
|
CH3OH + ClO reaction (R1, R2).
The equilibrium geometry of CH3OH is Cs symmetry and it has two types of non-equivalent H-atoms. Two transition states CH3O⋯H⋯OCl (TS11) and ClO⋯H⋯CH2OH (TS12) corresponds to the reaction path R1 and R2 (Fig. 2) respectively were found along the H-abstraction reaction of CH3OH with ClO radical. These two reactions occur by the participation of oxygen atom in ClO radical with the two non-equivalent hydrogen atoms of CH3OH. The methyl group orientation results in only one type of transition state TS12 for either syn or anti H-abstraction of the methyl group with respect to hydroxyl group orientation. From Table 2, it is observed that the two transition states, TS11 and TS12, are characterized by imaginary frequencies of 1946i cm−1 and 1250i cm−1 respectively. These imaginary frequencies correspond to the vibration of the molecule along its reaction pathway. The movement of hydrogen atom between O1 and O2 in TS11 and between C and O2 in TS12 leads to the product formation. Table 3 shows the electron density, ρ of different chemical bonds in TS11 and TS12. The ρ value for O1–H0 bond breaking and H0–O2 bond formation in TS11 are 0.176 au and 0.202 au respectively, whereas in TS12, breaking C–H1 and formation of H1–O2 bonds has electron density values of 0.196 au and 0.131 au respectively. The bonding analysis also shows the presence of other intermolecular bonds namely, H-bonding of Cl–H1 in TS11 and halogen bonding33,34 of Cl–O1 in TS12. The L-parameter35 which is the ratio of the increase in the bond length being broken over the increase in the bond length being formed is helpful in finding whether the transition state has reactant-like or product-like characteristics. The value of L more than 1 represents a product like TS and less than 1 represents reactant like TS. From Table 1, it is observed that for the reaction CH3OH + ClO, the estimated L value is more than 1 along the reaction path R1 and is less than 1 along the path R2. Thus, TS11 is product-like transition state and TS12 is reactant-like transition state. In TS11, O1–H0 bond increases by 24% (= 0.226 Å) compared to O1–H0 bond in the isolated CH3OH and H0–O2 bond increases by 19% (= 0.183 Å) when compared to H0–O2 bond in HOCl product molecule. On the other hand, in TS12, the elongation of C–H1 bond is about 13% (= 0.138 Å) compared to C–H1 bond in the isolated CH3OH and H1–O2 bond is stretched 38% (= 0.364 Å) when compared to H1–O2 bond in HOCl molecule. There is a slight shortening of C–O1 bond distance in both TS11 and TS12 than isolated CH3OH molecule. Table 1 shows that the bond angle θ(O1–H0–O2) is 165.6° in TS11 and thus attack of ClO on hydroxyl H-atom of CH3OH is distorted by 14.4° from collinearity. The bond angle θ(O2–H1–C) is 166.4° in TS12 which signifies that the attack of ClO on methyl H-atom is not collinear.
R1 leads to the formation of methoxy radical by the H-abstraction from the hydroxyl group of CH3OH whereas R2 path gives hydroxymethyl radical results from the methyl group H-abstraction of CH3OH. These two reaction pathways are associated with the formation of H-bonded pre- and post-reactive molecular complex on the potential energy surface connected by the transition states. These molecular complexes are characterized by all real frequencies (NIMAG = 0). Table 3 displays the different intra and intermolecular bond density present in these molecular complexes which are favored by the formation of intermolecular H-bonding or halogen bonding. The pre-reactive complex CH3OH⋯OCl (PreM11) and post-reactive complex CH3O⋯HOCl (PostM11) are connected by TS11. PreM11 is formed by the weak interaction of O-atom of ClO radical and hydroxyl H-atom of CH3OH. The complex PostM11 is formed between the O-atom of CH3O radical and H-atom of HOCl molecule. In PreM11, the electron density, ρ of intermolecular H0–O2 bond is 0.016 au and in PostM11, O1–H0 has bond density of 0.033 au. The weak intermolecular H0–O2 bond distance is 2.157 Å in PreM11 and O1–H0 distance is 1.845 Å in PostM11. In PostM11, there is another intermolecular H-bond, Cl–H1 of density 0.006 au and of distance 2.951 Å. This Cl–H1 bond is also present in TS11 with density 0.011 au and distance 2.645 Å. The bond angle θ(O1–H0–O2) is 141° and 157.2° in PreM11 and PostM11 respectively, which reflects that the atoms involved in bond breaking and forming are not linear in the weak complexes following R1 path. The dihedral angle ϕ(Cl–O2–H0–O1) is found to be −32.0° in PreM11 and it is −51.5° in PostM11. ClO approaches the methyl H-atom of CH3OH and forms the weak complex ClO⋯HCH2OH (PreM12) while complex ClOH⋯CH2OH (PostM12) is formed after the H-abstraction process following R2 reaction path as shown in Fig. 2. PreM12 results from the weak intermolecular interaction of oxygen atom of ClO with the methyl H-atom of CH3OH and PostM12 is formed from the weak interaction of H-atom of already formed HOCl with C-atom of CH2OH radical for R2 pathway. The newly formed weak intermolecular H1–O2 bond distance is 2.593 Å in PreM12 and C–H1 bond distance is 2.131 Å in PostM12. In PreM12, H1–O2 bond has density of 0.008 au and in PostM12, electron density for C–H1 bond is 0.022 au. PostM12 contains another intermolecular H-bond Cl–H0 with density of 0.012 au and distance of 2.607 Å. The halogen bond Cl–O1 has density 0.010 au in both PreM12 and TS12. The Cl–O1 bond distance is 3.112 Å in PreM12 and is 3.149 Å in TS12. At M06-2X/cc-pVTZ, the estimated dihedral angle ϕ(Cl–O2–H1–C) is 50.2° and 12.6° in PreM12 and PostM12 respectively. For R2 reaction pathway, the bond angle θ(O2–H1–C) is determined to be 107.9° in PreM12 and 152.0° in PostM12.
CH3OH + BrO reaction (R3, R4).
The optimized geometric parameters of all the species in the potential energy surface of the CH3OH + BrO reaction are given in Table 1. The attack of BrO radical on the hydroxyl H-abstraction (R3) corresponds to transition state, CH3O⋯H⋯OBr (TS21) and attack on the methyl H-abstraction (R4) corresponds to the transition state, BrO⋯H⋯CH2OH (TS22). The respective potential energy surface for R3 and R4 pathways are shown in Fig. 3 and corresponding bond breaking and bond forming densities are reported in Table 3. From the listed value in Table 3, it is observed that in TS21, electron density, ρ at BCP of O1–H0 bond breaking is lesser (= 0.176 au) than the corresponding ρ (= 0.372 au) in isolated CH3OH molecule and for the formation H0–O2 bonds ρ is lesser (= 0.196 au) than corresponding bond in the product HOBr molecule (= 0.366 au). In TS22, C–H1 density of bond breaking is 0.201 au and density of H1–O2 bond formation is 0.126 au. The second intermolecular H-bond Br–H1 in TS21 has density of 0.015 au and distance of 2.630 Å. As reported in Table 2, imaginary frequency associated with the O1⋯H0⋯O2 vibration in TS21 is 1918i cm−1 and with C⋯H1⋯O2 vibration, it is 1124i cm−1 in TS22. In case of both ClO and BrO radicals, the magnitude of the imaginary frequency for the transition state involving methyl H-abstraction is less than the hydroxyl H-abstraction path. The L value for TS21 is more than 1 and for TS22, it is less than 1. These L values indicate that TS21 is product like whereas TS22 geometry is reactant like. The characteristics of TS21 and TS22 are similar to the transition states TS11 and TS12 for the H-abstraction reaction from CH3OH by ClO radical. At M06-2X/cc-pVTZ, the O1–H0 bond is extended by 0.229 Å (= 24%) in TS21 when compared to O1–H0 bond in isolated CH3OH molecule and the bond length H0–O2 is greater by 0.192 Å (= 20%) from the HOCl molecule. The C–H1 and H1–O2 bonds in TS22 at M06-2X/cc-pVTZ are 1.224 Å and 1.341 Å respectively. Thus in TS22, the increase in C–H1 bond is 0.130 Å (= 12%) than C–H1 bond in the isolated CH3OH molecule and elongation in forming H1–O2 bond is by 0.377 Å (= 39%) comparative to H1–O2 bond in HOCl molecule. The extent of C–O1 bond shortening in TS21 and TS22 is similar as in TS11 and TS12. The bond angle θ(O1–H0–O2) is 164.7° in TS21 and θ(O2–H1–C) is 168.3° in TS22 were obtained at M06-2X/cc-pVTZ. These bond angles reflect that the attack of BrO on hydroxyl H-atom or on methyl H-atom of CH3OH is not collinear.
From the potential energy surface of CH3OH + BrO reaction, it is noticed that the TS is connected to the loosely bound PreM in the backward direction and PostM in the forward direction. It can be observed from the bonding density in Table 3, that the molecular complexes associated with CH3OH + BrO reaction are driven by the formation of intermolecular H-bonding and halogen bonding. TS21 is the saddle point with pre-reactive complex CH3OH⋯OBr (PreM21) and post-reactive complex CH3O⋯HOBr (PostM21) for R3 reaction pathway while TS22 is connected to BrO⋯HCH2OH (PreM22) and BrOH⋯CH2OH (PostM12) for R4 pathway. Geometric parameters of the PreMs are close to CH3OH and BrO and parameters for PostMs are related to isolated HOCl and CH3O radical (PostM21) or CH2OH radical (PostM22). At the entrance channel, the intermolecular distance H0–O2 is 2.115 Å in PreM21 and H1–O2 distance is 2.569 Å in PreM22 as obtained at M06-2X/cc-pVTZ. On the other hand, at the exit channel, the intermolecular O1–H0 bond distance is 1.862 Å in PostM21 and C–H1 bond distance is 2.150 Å in PostM22. The H0–O2 bond density is 0.018 au in PreM21 and H1–O2 density is 0.008 au in PreM22. The second intermolecular Br–O1 bond distance is 3.217 Å in PreM22 and is 3.253 Å in TS22 and both stationary points have density of 0.010 au. O1–H0 bond density is again found to be 0.032 au in PostM21 and C–H1 bond density is 0.021 au in PostM22. In PostM22, second H-bond, Br–H0 is present with distance 2.751 Å and density 0.011 au. At M06-2X/cc-pVTZ, level of theory the calculated bond angle θ(O1–H0–O2) for PreM21 and PostM21 are 143.8° and 156.6° respectively, and θ(O2–H1–C) for PreM22 is 107.9° and for PostM22 is 153.5°. The dihedral angle ϕ(Br–O2–H0–O1) for the molecular complexes following R3 path are 30.6° in PreM21 complex and is 54.3° in PostM21 complex. For the reaction CH3OH + BrO following R4 path, the dihedral angle ϕ(Br–O2–H1–C) is 52.1° for the pre-reactive molecular complex PreM22 and 15.2° for the post-reactive molecular complex PostM22.
3.2. Energetics
The different kinetic and thermodynamic parameters for all the species involved in CH3OH + ClO (R1, R2) and CH3OH + BrO (R3, R4) reactions at CCSD(T)/cc-pVTZ//M06-2X/cc-pVTZ are given in Tables 4 and 5. All these parameters are ZPE and spin–orbit corrected. Table 4 includes the adiabatic barrier height (ΔE#0), enthalpy of activation (ΔH#0), Gibbs free energy of activation (ΔG#0), enthalpy of reaction (ΔrH0), reaction free energies (ΔrG0), entropy (ΔS0) and total potential energies (ΔrE0) of products relative to the reactants in the reaction of CH3OH with XO (X = Cl, Br) radical calculated at 298.15 K. The reaction free energies of the hydroxyl H-abstraction reaction from CH3OH by both ClO (R1) and BrO (R3) radicals show that these pathways are endergonic reaction (ΔG > 0). In Table 4, the calculated free energies of the reactions proceeding through R2 and R4 pathways for the methyl H-abstraction reaction from CH3OH by ClO and BrO radicals are −0.47 kcal per mol and −1.60 kcal per mol and thus these are exergonic reaction (ΔG < 0). At 298.15 K, reactions following R1, R2 pathways with ClO radical are endothermic along with the R3 reaction pathway with BrO radical (ΔH > 0). The reaction enthalpy for the methyl H-abstraction by BrO radical (R4) calculated at CCSD(T)/cc-pVTZ//M06-2X/cc-pVTZ is exothermic by a slight negative value of 0.47 kcal per mol. The adiabatic barrier height, ΔE#0 calculated at CCSD(T)/cc-pVTZ//M06-2X/cc-pVTZ for all the pathways R1, R2, R3 and R4 including spin–orbit and ZPE correction are reported in Table 4. This barrier height is found to be positive for both the possible H-abstraction pathways with ClO and BrO radicals. In Table 4, CCSD(T)/cc-pVTZ//M06-2X/cc-pVTZ computed barrier height relative to reactants are 13.31 kcal per mol, 8.81 kcal per mol, 12.52 kcal per mol and 8.35 kcal per mol for R1, R2, R3 and R4 respectively. From these barrier heights it may be gleaned that the reactions for hydroxyl H-abstraction (R1, R3) have higher barrier height than the methyl H-abstraction pathways (R2, R4). Table 5 reports the kinetic and thermodynamic parameters for the pre-reactive molecular complexes (PreM11, PreM12, PreM21, PreM22) and post-reactive molecular complexes (PostM11, PostM12, PostM21, PostM22) computed at CCSD(T)/cc-pVTZ//M06-2X/cc-pVTZ and 298.15 K. The XO (X = Cl, Br) radical approaches CH3OH for H-abstraction from hydroxyl or methyl group and forms weak bonded intermolecular PreM complexes which are more stabilized than the reactants by an amount of 2.06 kcal per mol (PreM11), 1.11 kcal per mol (PreM12), 3.16 kcal per mol (PreM21) and 1.59 kcal per mol (PreM22). Post-reactive molecular complexes for R1 (PostM11) and R3 (PostM21) reactions show less stability when compared to reactants and for R3 (PostM12) and R4 (PostM22) are more stable than the reactants. At CCSD(T)/cc-pVTZ//M06-2X/cc-pVTZ all the post-reactive complexes on each potential energy surfaces are more stable than the products. From Fig. 2 and 3, it is observed that the TSs are above the pre-reactive and post-reactive molecular complexes on the potential energy surface for all pathways.
Table 4 Kinetic and thermodynamic parameters (in kcal per mol) of the transition states and the overall reaction for H-abstraction reaction of CH3OH by XO (X = Cl, Br) radical relative to reactants at 298.15 K. All values are spin–orbit and ZPE corrected
| Reaction |
CCSD(T)/cc-pVTZ//M06-2X/cc-pVTZ |
| ΔE#0 |
ΔG#0 |
ΔH#0 |
ΔrE0 |
ΔrG0 |
ΔrH0 |
ΔS0 |
| CH3OH + OCl → CH3O + HOCl (R1) |
13.31 |
21.92 |
12.75 |
7.57 |
6.55 |
7.90 |
4.53 |
| CH3OH + OCl → CH2OH + HOCl (R2) |
8.81 |
17.38 |
8.14 |
0.38 |
−0.47 |
0.66 |
3.79 |
| CH3OH + OBr → CH3O + HOBr (R3) |
12.52 |
21.23 |
11.96 |
6.43 |
5.41 |
6.76 |
4.53 |
| CH3OH + OBr → CH2OH + HOBr (R4) |
8.35 |
17.50 |
7.63 |
−0.76 |
−1.60 |
−0.47 |
3.79 |
Table 5 Kinetic and thermodynamic parameters (in kcal per mol) of the pre- and post-reactive molecular complexes for H-abstraction reaction of CH3OH with XO (X = Cl, Br) radical relative to reactants at 298.15 K. All values are spin–orbit and ZPE corrected
| Species |
CCSD(T)/cc-pVTZ//M06-2X/cc-pVTZ |
| ΔE0 |
ΔG0 |
ΔH0 |
| PreM11 |
−2.06 |
5.43 |
−2.06 |
| PreM12 |
−1.11 |
5.54 |
−0.81 |
| PreM21 |
−3.16 |
4.06 |
−3.03 |
| PreM22 |
−1.59 |
5.33 |
−1.31 |
| PostM11 |
2.61 |
9.73 |
2.65 |
| PostM12 |
−3.93 |
4.14 |
−3.91 |
| PostM21 |
1.63 |
9.12 |
1.66 |
| PostM22 |
−5.13 |
2.92 |
−5.04 |
3.3. CH3OH + XO (X = Cl, Br) reaction kinetics
Table 6 reports the fitted rate constant data at CCSD(T)/cc-pVTZ//M06-2X/cc-pVTZ for CH3OH + ClO (kR1, kR2) and CH3OH + BrO (kR3, kR4) reactions computed at different temperature within the range 200–2500 K. From Table 6, it can be observed that the rate constants, kR2 and kR4 for methyl H-abstraction from CH3OH leading to hydroxymethyl (CH2OH) radical formation shows larger rate constant than rate constants, kR1 and kR3 for hydroxyl H-abstraction forming methoxy (CH3O) radical for the studied temperature for both ClO and BrO radicals. At lower temperatures (<1000 K) formation of CH3O radical from CH3OH is negligible for both ClO and BrO radicals which is more pronounced in case of ClO radical. Due to the presence of upward curvature, the calculated CVT rate constants were fitted to the modified three parameter Arrhenius equation of the type k(T) = ATnexp(−Ea/RT) rather than two parameter Arrhenius equation and the plots are shown in Fig. 4 and in Table 7, the fitted Arrhenius parameters A, n and Ea for all four reactions: R1, R2, R3, R4 are given. The fitted Arrhenius equations in units of cm3 per molecule per s for CH3OH + XO (X = Cl, Br) reactions are
| kR1(T) = 4.55 × 10−20T1.70exp(−4091/T) |
| kR2(T) = 7.37 × 10−19T1.54exp(−2097/T) |
| kR3(T) = 7.39 × 10−23T2.74exp(−2093/T) |
| kR4(T) = 1.74 × 10−20T2.38exp(−2217/T) |
| kov1(T) = 3.92 × 10−19T1.63exp(−2062/T) |
| kov2(T) = 1.53 × 10−20T2.41exp(−2206/T) |
Table 6 CVT calculated rate constants (cm3 per molecule per s) and branching ratio (ϕ in %) of CH3OH + ClO (R1, R2) and CH3OH + BrO (R3, R4) reactions for the temperature range 200–2500 K at CCSD(T)/cc-pVTZ//M06-2X/cc-pVTZ
| Temperature (K) |
Rate constant, k (cm3 per molecule per s) |
Branching ratio, ϕ (%) |
|
k
R1
|
k
R2
|
k
ov1
|
k
R3
|
k
R4
|
k
ov2
|
R1
|
R2
|
R3
|
R4
|
| 200 |
4.84 × 10−25 |
7.25 × 10−20 |
7.25 × 10−20 |
4.25 × 10−21 |
8.20 × 10−20 |
8.62 × 10−20 |
0.00 |
100.00 |
4.93 |
95.07 |
| 220 |
3.65 × 10−24 |
2.18 × 10−19 |
2.18 × 10−19 |
1.43 × 10−20 |
2.82 × 10−19 |
2.96 × 10−19 |
0.00 |
100.00 |
4.82 |
95.18 |
| 240 |
1.99 × 10−23 |
5.51 × 10−19 |
5.51 × 10−19 |
4.00 × 10−20 |
8.03 × 10−19 |
8.43 × 10−19 |
0.00 |
100.00 |
4.75 |
95.25 |
| 260 |
8.47 × 10−23 |
1.22 × 10−18 |
1.22 × 10−18 |
9.74 × 10−20 |
1.98 × 10−18 |
2.07 × 10−18 |
0.01 |
99.99 |
4.69 |
95.31 |
| 280 |
2.95 × 10−22 |
2.43 × 10−18 |
2.43 × 10−18 |
2.12 × 10−19 |
4.34 × 10−18 |
4.55 × 10−18 |
0.01 |
99.99 |
4.66 |
95.34 |
| 298 |
7.93 × 10−22 |
4.21 × 10−18 |
4.21 × 10−18 |
3.95 × 10−19 |
8.12 × 10−18 |
8.51 × 10−18 |
0.02 |
99.98 |
4.64 |
95.36 |
| 320 |
2.30 × 10−21 |
7.62 × 10−18 |
7.62 × 10−18 |
7.78 × 10−19 |
1.60 × 10−17 |
1.68 × 10−17 |
0.03 |
99.97 |
4.62 |
95.38 |
| 340 |
5.41 × 10−21 |
1.23 × 10−17 |
1.23 × 10−17 |
1.35 × 10−18 |
2.79 × 10−17 |
2.92 × 10−17 |
0.04 |
99.96 |
4.62 |
95.38 |
| 360 |
1.16 × 10−20 |
1.89 × 10−17 |
1.89 × 10−17 |
2.22 × 10−18 |
4.59 × 10−17 |
4.81 × 10−17 |
0.06 |
99.94 |
4.62 |
95.38 |
| 380 |
2.32 × 10−20 |
2.79 × 10−17 |
2.80 × 10−17 |
3.50 × 10−18 |
7.21 × 10−17 |
7.56 × 10−17 |
0.08 |
99.92 |
4.62 |
95.38 |
| 400 |
4.33 × 10−20 |
3.98 × 10−17 |
3.99 × 10−17 |
5.30 × 10−18 |
1.09 × 10−16 |
1.14 × 10−16 |
0.11 |
99.89 |
4.63 |
95.37 |
| 500 |
4.89 × 10−19 |
1.60 × 10−16 |
1.61 × 10−16 |
2.78 × 10−17 |
5.63 × 10−16 |
5.90 × 10−16 |
0.30 |
99.70 |
4.71 |
95.29 |
| 600 |
2.61 × 10−18 |
4.27 × 10−16 |
4.30 × 10−16 |
9.19 × 10−17 |
1.82 × 10−15 |
1.91 × 10−15 |
0.61 |
99.39 |
4.81 |
95.19 |
| 700 |
8.97 × 10−18 |
8.92 × 10−16 |
9.01 × 10−16 |
2.31 × 10−16 |
4.45 × 10−15 |
4.68 × 10−15 |
1.00 |
99.00 |
4.93 |
95.07 |
| 800 |
2.33 × 10−17 |
1.59 × 10−15 |
1.62 × 10−15 |
4.83 × 10−16 |
9.10 × 10−15 |
9.58 × 10−15 |
1.44 |
98.56 |
5.05 |
94.95 |
| 900 |
5.03 × 10−17 |
2.56 × 10−15 |
2.61 × 10−15 |
8.92 × 10−16 |
1.64 × 10−14 |
1.73 × 10−14 |
1.93 |
98.07 |
5.16 |
94.84 |
| 1000 |
9.48 × 10−17 |
3.79 × 10−15 |
3.89 × 10−15 |
1.50 × 10−15 |
2.70 × 10−14 |
2.85 × 10−14 |
2.44 |
97.56 |
5.28 |
94.72 |
| 1100 |
1.62 × 10−16 |
5.32 × 10−15 |
5.48 × 10−15 |
2.36 × 10−15 |
4.14 × 10−14 |
4.37 × 10−14 |
2.95 |
97.05 |
5.39 |
94.61 |
| 1200 |
2.56 × 10−16 |
7.13 × 10−15 |
7.38 × 10−15 |
3.51 × 10−15 |
6.02 × 10−14 |
6.37 × 10−14 |
3.46 |
96.54 |
5.51 |
94.49 |
| 1300 |
3.81 × 10−16 |
9.22 × 10−15 |
9.60 × 10−15 |
5.00 × 10−15 |
8.40 × 10−14 |
8.90 × 10−14 |
3.96 |
96.04 |
5.61 |
94.39 |
| 1400 |
5.40 × 10−16 |
1.16 × 10−14 |
1.21 × 10−14 |
6.87 × 10−15 |
1.13 × 10−13 |
1.20 × 10−13 |
4.45 |
95.55 |
5.72 |
94.28 |
| 1500 |
7.38 × 10−16 |
1.43 × 10−14 |
1.50 × 10−14 |
9.16 × 10−15 |
1.48 × 10−13 |
1.58 × 10−13 |
4.92 |
95.08 |
5.82 |
94.18 |
| 1600 |
9.77 × 10−16 |
1.72 × 10−14 |
1.82 × 10−14 |
1.19 × 10−14 |
1.90 × 10−13 |
2.02 × 10−13 |
5.38 |
94.62 |
5.91 |
94.09 |
| 1800 |
1.58 × 10−15 |
2.38 × 10−14 |
2.54 × 10−14 |
1.90 × 10−14 |
2.93 × 10−13 |
3.12 × 10−13 |
6.24 |
93.76 |
6.10 |
93.90 |
| 2000 |
2.38 × 10−15 |
3.15 × 10−14 |
3.39 × 10−14 |
2.86 × 10−14 |
4.26 × 10−13 |
4.55 × 10−13 |
7.03 |
92.97 |
6.28 |
93.72 |
| 2500 |
5.23 × 10−15 |
5.48 × 10−14 |
6.00 × 10−14 |
6.49 × 10−14 |
9.06 × 10−13 |
9.71 × 10−13 |
8.72 |
91.28 |
6.68 |
93.32 |
 |
| | Fig. 4 Arrhenius plot for the temperature range 200–2500 K fitted to the calculated CVT rate constants for the two pathways of (a) CH3OH + ClO (R1, R2) and (b) CH3OH + BrO (R3, R4) reactions. | |
Table 7 Calculated Arrhenius parameters over the temperature range 200–2500 K for CH3OH + XO (X = Cl, Br) reaction
| Reaction |
CCSD(T)/cc-pVTZ//M06-2X/cc-pVTZ |
|
A (cm3 per molecule per s) |
n
|
E
a (kcal per mol) |
| CH3OH + OCl → CH3O + HOCl (R1) |
4.55 × 10−20 |
1.70 |
8.13 |
| CH3OH + OCl → CH2OH + HOCl (R2) |
7.37 × 10−19 |
1.54 |
4.17 |
| CH3OH + OBr → CH3O + HOBr (R3) |
7.39 × 10−23 |
2.74 |
4.16 |
| CH3OH + OBr → CH2OH + HOBr (R4) |
1.74 × 10−20 |
2.38 |
4.40 |
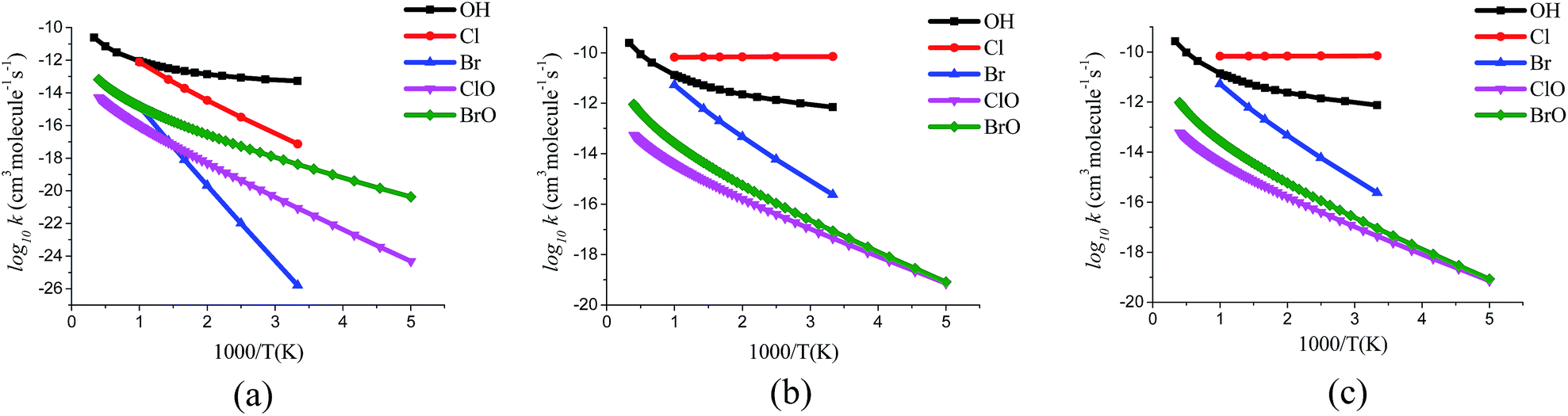
In CH3OH, the C–H bond strength (= 94 kcal per mol) is lower than the O–H bond strength (= 104 kcal per mol).36 For the temperature range under investigation, the H-abstraction from methyl group is dominated over the H-abstraction from hydroxyl group of CH3OH by ClO and BrO radicals. The overall rate constant kov1 (= kR1 + kR2) and kov2 (= kR3+ kR4) are also reported in Table 6. At higher temperatures (>1000 K), hydroxyl H-abstraction reaction has some contribution towards the overall rate constants. The branching ratio (ϕ), which is defined as the ratio for a particular pathway rate constant divided by the sum of rate constants for all parallel reaction pathways are also included in Table 6 and the plots are shown in Fig. 5. These branching ratios are used to emphasize the importance of one pathway over the other one. It is necessary to know the branching ratio which is the indicator of preference of a pathway at different temperatures. At low temperature, the branching ratio for the reaction formation of CH2OH product radical is 100% with ClO radical. As the temperature increases, the reaction pathway contribution from CH3O product-radical formation reaction also increases. With the BrO radical, the branching ratio value for CH2OH product radical formation is found to be more than 90% at all temperatures for the entire temperature range studied. We have compared our results of studied reaction with the H-abstraction reaction from CH3OH by OH, Cl and Br radicals available in literature. For the H-abstraction reaction from CH3OH by hydroxyl (OH) radical, the rate constant following methyl H-abstraction was found to be dominant at low temperature over the hydroxyl H-abstraction reaction.37,38 As temperature rises, the hydroxyl H-abstraction rate constant also becomes important. This is also consistent with the H-abstraction reaction of CH3OH + Cl but for the reaction CH3OH + Br, the rate constant for methoxy radical formation pathway is found to be inactive even at higher temperature.2,39 Our results for H-abstraction with ClO and BrO radicals are also similar to the reaction with OH, Cl, Br radical where hydroxymethyl radical forms predominantly. A comparison plot for the reactivity of OH, Cl, Br, ClO and BrO for the H-abstraction from CH3OH is shown in Fig. 6. Among these radicals in Fig. 6, it is observed that Cl radical shows the highest rate constant for the methyl H-abstraction from CH3OH and thus the overall rate constant. The hydroxyl H-abstraction rate constant is found to be more for OH than other radicals but the overall rate constant for OH is lower than Cl radical. It is because of higher contribution from methyl H-abstraction to the overall by Cl than OH. Cl forms barrierless transition state2 for methyl H-abstraction and thus contributes more to the overall rate constant than OH. Kinetically, H-abstraction from CH3OH by Cl or Br radicals are more favourable than XO (X = Cl, Br) radicals where H-abstraction involved O-atom of XO radicals. Substitution of H-atom of OH radical by halogen atoms makes it less reactive than either OH or X radicals. Where Cl shows larger rate of H-abstraction reaction than Br radical, ClO has lower rate constant value than BrO over the studied temperature range. For methyl H-abstraction reaction and overall rate of the reaction, CH3OH + Cl shows a noticeable difference from CH3OH + Br reaction. At lower temperature this difference is higher and as temperature increases the difference becomes smaller. From Fig. 6, it is observed that the methyl H-abstraction and overall rate of the reaction shows smaller difference at lower temperatures in case of reaction of ClO and BrO when compared to the reaction of Cl and Br. Temperature has less effect on methyl H-abstraction by Cl radical due to the barrierless transition state but has more effect on the reaction of OH, Br, ClO and BrO radicals. Indeed, as temperature increases there is an increase in rate also. Experimental study on CH3OH + XO (X = Cl, Br) is not known, there is available experimental data for the H-abstraction reaction from CH3OH by OH or halogen radicals using different experimental techniques like fast flow or laser photolysis-resonance fluorescence.37,39,40 The experimental study of CH3OH + XO (X = Cl, Br) reaction can be attempted using these techniques and the present study will be helpful for analyzing the branching ratio of methoxy or hydroxymethyl radical formation at different temperature.
 |
| | Fig. 5 Plot of branching ratios (ϕ in %) versus temperature (T) for the two pathways of (a) CH3OH + ClO (R1, R2) and (b) CH3OH + BrO (R3, R4) reactions. | |
 |
| | Fig. 6 Arrhenius plot for the rate constants of (a) hydroxyl H-abstraction (b) methyl H-abstraction (c) overall H-abstraction reactions from CH3OH by OH, Cl, Br, ClO and BrO radicals. Rate constant data for OH are taken from ref. 38 and for Cl, Br radicals it is taken from ref. 2. | |
4. Conclusion
The kinetic and mechanism study of the two channel H-abstraction reaction of CH3OH + XO (X = Cl, Br) was performed with the use of M06-2X and CCSD(T) level of theory combined with cc-pVTZ basis set. Pre-reactive and post-reactive molecular complexes were located at entrance and exit channel, respectively for CH3OH + ClO → CH3O + HOCl (R1), CH3OH + ClO → CH2OH + HOCl (R2), CH3OH + BrO → CH3O + HOBr (R3) and CH3OH + BrO → CH2OH + HOBr (R4) reactions. The potential energy surface of R1, R2, R3 and R4 reactions were obtained at CCSD(T)/cc-pVTZ//M06-2X/cc-pVTZ. The rate constants for all the reactions were calculated using canonical variational transition state theory with unsymmetrical Eckart tunneling correction. The calculated rate constants were fitted to modified three parameters Arrhenius equation for all the reaction pathways. At CCSD(T)/cc-pVTZ//M06-2X/cc-pVTZ, the rate constant associated with the hydroxymethyl radical formation was found to be higher than the methoxy radical formation for both ClO and BrO radicals for the entire temperature range of 200–2500 K. The importance of formation of one product over the other was analyzed by calculating the branching ratio.
Acknowledgements
S.B. is thankful to Indian Institute of Technology Patna for financial support and for providing research facilities at IIT Patna.
References
- T. S. Norton and F. L. Dryer, Int. J. Chem. Kinet., 1990, 22, 219–241 CrossRef CAS.
- J. T. Jodkowski, M.-T. Rayez, J.-C. Rayez, T. Bérces and S. Dóbé, J. Phys. Chem. A, 1998, 102, 9230–9243 CrossRef CAS.
- D. J. Jacob, B. D. Field, Q. Li, D. R. Blake, J. de Gouw, C. Warneke, A. Hansel, A. Wisthaler, H. B. Singh and A. Guenther, J. Geophys. Res.: Atmos., 2005, 110, D08303 Search PubMed.
- A. H. Goldstein and I. E. Galbally, Environ. Sci. Technol., 2007, 41, 1514–1521 CrossRef CAS PubMed.
- R. C. Macdonald and R. Fall, Atmos. Environ., 1993, 27, 1709–1713 CrossRef.
- S. Solomon, R. R. Garcia, F. S. Rowland and D. J. Wuebbles, Nature, 1986, 321, 755–758 CrossRef CAS.
- J. Gonzalez, J. M. Anglada, R. J. Buszek and J. S. Francisco, J. Am. Chem. Soc., 2011, 133, 3345–3353 CrossRef CAS PubMed.
- Y. Bedjanian and G. Poulet, Chem. Rev., 2003, 103, 4639–4655 CrossRef CAS PubMed.
- P. W. M. Jacobs and H. M. Whitehead, Chem. Rev., 1969, 69, 551–590 CrossRef CAS.
- S. Yuasa, S. Yushina, T. Uchida and N. Shiraishi, Proc. Combust. Inst., 2000, 28, 863–870 CrossRef CAS.
- H. C. Urey and H. Johnston, Phys. Rev., 1931, 38, 2131–2152 CrossRef CAS.
- E. H. Coleman and A. G. Gaydon, Discuss. Faraday Soc., 1947, 2, 166–169 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- G. Paytakov, T. Dinadayalane and J. Leszczynski, J. Phys. Chem. A, 2015, 119, 1190–1200 CrossRef CAS PubMed.
- X. Xu, I. M. Alecu and D. G. Truhlar, J. Chem. Theory Comput., 2011, 7, 1667–1676 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
-
R. Dennington, T. Keith and J. Millam, GaussView, Version 5, Semichem Inc., Shawnee Mission KS, 2009 Search PubMed.
- A. Fernandez-Ramos, B. A. Ellingson, B. C. Garrett and D. G. Truhlar, Rev. Comput. Chem., 2007, 23, 125–232 CAS.
- C. Eckart, Phys. Rev., 1930, 35, 1303–1309 CrossRef CAS.
- S. Canneaux, F. Bohr and E. Hénon, J. Comput. Chem., 2014, 35, 82–93 CrossRef CAS PubMed.
- B. C. Garrett and D. G. Truhlar, J. Phys. Chem., 1979, 83, 1079–1112 CrossRef CAS.
- S. Begum and R. Subramanian, J. Mol. Model., 2014, 20, 2262 CrossRef PubMed.
- S. Canneaux, B. Xerri, F. Louis and L. Cantrel, J. Phys. Chem. A, 2010, 114, 9270–9288 CrossRef CAS PubMed.
- S.-H. Lee and K. Liu, J. Chem. Phys., 1999, 111, 6253–6259 CrossRef CAS.
- B. J. Drouin, C. E. Miller, E. A. Cohen, G. Wagner and M. Birk, J. Mol. Spectrosc., 2001, 207, 4–9 CrossRef CAS PubMed.
- B. J. Drouin, C. E. Miller, H. S. P. Muller and E. A. Cohen, J. Mol. Spectrosc., 2001, 205, 128–138 CrossRef CAS PubMed.
-
R. F. W. Bader, Atoms in Molecules. A Quantum Theory, Clarendon Press, Oxford, England, 1990 Search PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
-
T. A. Keith, AIMALL, version 13.11.04, TK Gristmill Software, Overland Park, KS, USA, 2013, http://aim.tkgristmill.com Search PubMed.
- P. Politzer, P. Lane, M. C. Concha, Y. Ma and J. S. Murray, J. Mol. Model., 2007, 13, 305–311 CrossRef CAS PubMed.
- P. Politzer and J. S. Murray, ChemPhysChem, 2013, 14, 278–294 CrossRef CAS PubMed.
- M. T. Rayez, J. C. Rayez and J. P. Sawerysyn, J. Phys. Chem., 1994, 98, 11342–11352 CrossRef CAS.
- D. F. McMillen and D. M. Golden, Annu. Rev. Phys. Chem., 1982, 33, 493–532 CrossRef CAS.
- W. P. Hesst and F. P. Tully, J. Phys. Chem., 1989, 93, 1944–1947 CrossRef.
- J. T. Jodkowski, M.-T. Rayez, J.-C. Rayez, T. Bérces and S. Dóbé, J. Phys. Chem. A, 1999, 103, 3750–3765 CrossRef CAS.
- A. Garzón, C. A. Cuevas, A. A. Ceacero, A. Notario, J. Albaladejo and M. Fernández-Gómez, J. Chem. Phys., 2006, 125, 104305 CrossRef PubMed.
- S. Dóbé, T. Bérces, T. Turanyi, F. Marta, J. Grussdorf, F. Temps and H. G. Wagner, J. Phys. Chem. A, 1996, 100, 19864–19873 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Structural parameters and Cartesian coordinates for optimized geometries of reactants, transition states, molecular complexes and products at M06-2X/cc-pVTZ level of theory. See DOI: 10.1039/c5ra06483j |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a