
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular mechanisms for the adhesion of chitin and chitosan to montmorillonite clay†
Yan
Wang
a,
Jakob
Wohlert
*bc,
Malin
Bergenstråhle-Wohlert
bc,
Yaoquan
Tu
a and
Hans
Ågren
*a
aDivision of Theoretical Chemistry and Biology, School of Biotechnology, KTH Royal Institute of Technology, SE-106 91 Stockholm, Sweden. E-mail: agren@theochem.kth.se
bDepartment of Fibre and Polymer Technology, School of Chemical Science and Engineering, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden. E-mail: jacke@kth.se
cWallenberg Wood Science Centre, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden
First published on 12th June 2015
Abstract
Molecular dynamics simulations have been performed to investigate molecular adhesion of chitin and chitosan oligomers to montmorillonite (Mnt) clay at different degrees of acetylation (DA, 0%, 20%, 40%, 60%, 80% and 100%) and different degree of protonation (DPr, 0%, 50%, 100% mimicking pH > 6.5, pH = 6.5, pH < 4, respectively) under fully hydrated conditions. Although the Mnt surface is negatively charged and a variation in DA also implies going from a positively charged oligomer at DA = 0% to a neutral oligomer at DA = 100%, the simulations show unexpectedly small variation of the total molecular adhesion as a function of DA. From our analysis we propose that this quantitatively similar adhesion arises from two different mechanisms. At low DA, the oligomer is rich in positively charged amino groups interacting strongly with the negatively charged surface by direct electrostatic interaction. On the other hand, at high DA, electrically neutral acetyl groups are strongly correlated with the Na+ counter ions, which are in all cases stuck at the surface and the counter ions seem to act as ‘glue’ between the acetyl groups and the Mnt. However, when protonation was decreased, adhesion was affected and significantly lowered at neutral conditions (DPr = 0%). The reason is concluded to be differences in charge distributions of the respective functional groups. A further investigation on the intramolecular hydrogen bonds formed in CHT or CHS shows that the adsorbed conformation of the polymer is also highly affected by DA. This work provides fundamental insights into adhesion mechanisms and is of potential importance for the development of polymer–clay based composite materials.
Introduction
The field of research on polymer–clay nanocomposite materials has been appealing to material developers since the mid 1980's.1 By adding just a few weight percent of montmorillonite (Mnt) clay to a polymer matrix, the final material possesses mechanical, thermal and gas barrier properties superior to those of the pure matrix.2–4 When using biopolymers, this class of material becomes interesting also from a sustainability perspective.Chitin (CHT) is the second most abundant naturally occurring polymer in the world, after cellulose.5,6 Structurally, CHT is a polysaccharide composed of β(1-4) linked glucosamine and N-acetylated glucosamine units. It is found in nature in the form of microfibrils, as the principal load-bearing component in the shells of crustaceans, the exoskeletons of arthropods, mushrooms and the cell walls of fungi.7 Despite its abundance, low cost and ‘green’ origin, industrial utilization of CHT is severely limited because of its poor solubility in most, including aqueous, solvents. This is possibly related to its highly acetylated structure.8 Chitosan (CHS), on the other hand which is the main derivative of CHT, possesses many of the advantageous traits of CHT and is, at the same time, soluble in acidic aqueous solutions. The gain in solubility is an effect of alkaline deacetylation of CHT, and the protonation of the amino groups in the acidic condition (pH < 6.5).8,9 Generally, the degree of acetylation (DA), which refers to the proportion of acetylated glucosamine units to the total number of units, should be lower than 50% for the polymer to qualify as chitosan.10 Chitosan is widely employed in food science, biomedical and cosmetic industries and agriculture,11 and in addition, being the only positively charged biopolymer at low pH, chitosan can be used as a flocculant to induce the deposition of anionic compounds12 and efficiently adsorb on an oppositely charged substrate to produce multilayered thin films from aqueous solutions.5,10
The idea of combining the bio-polymer CHS with the Mnt clay is useful for designing novel and renewable materials. There are various approaches to mimicking the brick-and-mortar structure of nacre.13 Tang et al.14 pioneered the field of nacre-mimetic composites with a layer-by-layer deposition technique using clay and a water-soluble polymer, and Walther et al.15,16 provided the next large step forward by utilizing the self-assembly properties of the two phases in a process similar to paper making, thereby permitting a continuous water-based processing. This approach was later extended to biopolymer matrices like xyloglucan17,18 and also chitosan.19 Humidity is known to significantly lower the mechanical properties of the final material,14 although the effect can be minimized either by chemical cross-linking17 or by using a polymer that has intrinsically high affinity for clay in water,18 and thus can provide good stress-transfer also at high moisture contents. In addition, the ability of chitosan to adsorb on the clay during processing is an important factor that governs the nanostructure.20 Hence, it is important to understand the driving forces at the interface within a fully hydrated polymer–Mnt system, which is the focus of the present study.
Since chitosan is positively charged, it will intuitively bind strongly to the negatively charged Mnt. However, the adsorption mechanism seems more complicated than being the effect of pure electrostatic attraction only. Experimentally, Tiraferri et al.10 have studied the chitosan adsorption mechanism on oppositely charged silica surfaces by combining optical reflectometry and quartz crystal microbalance techniques. They concluded that the adsorption behavior is highly dependent on the aqueous solubility of the polymer, which in turn is closely related to pH, and partially influenced by the electrolyte type and concentration used in the solution. In particular, at acidic pH, chitosan behaved similarly to weakly charged polyelectrolytes and the driving force of the adsorption was found to be electrostatic attraction and, possibly, counterion release. However, as the pH value increased, aggregates were formed and large amounts of water were trapped. However, their discussion is limited by the fact that the chitosan sample used in their study was characterized only by molecular weight and an average DA. Franca et al.8 used molecular dynamics (MD) simulations to show that the solvation is highly dependent on DA, and that the distribution of the acetylated groups and the protonated amino group affects a whole range of features of chitosan. Moreover, water mobility and the intramolecular interactions among chitosan chains seem to affect the stability and solubility as well.8,9,21
MD simulations has evolved into a suitable tool to deal with specific questions in material science, for instance, the interactions between organic polymer and clay minerals in aqueous solution.3,22,23 We have recently applied MD to study the adhesion of the polysaccharide xyloglucan (XG) to a Mnt clay surface.24,25 In the present study, we continue to focus on interfaces and more specifically, investigate how the degree of acetylation as well as protonation influences the polymer–clay interaction. To this end, extensive MD simulations have been performed, and the work of adhesion between polymer and clay is evaluated using steered MD simulations and umbrella sampling, at several DA's ranging from 0% to 100%, and for degrees of protonation (DPr) of 0%, 50%, and 100%. The respective roles of acetylation and protonation for polymer adsorption, conformation, and solubility are discussed. The simulations provide theoretical insight into the complex interfacial structure within a chitosan–clay nanocomposite material. The effect of acetylation and protonation upon adhesion is important to the community developing these materials from a processing but also moisture sensitivity perspective.
Methods
Theoretical model
The modelled polymer is a 10-mer polysaccharide chain (Fig. 1a) consisting of 2, 4, 6, 8 or 10 acetylated glucosamine units corresponding to a DA of 20%, 40%, 60%, 80% and 100%, respectively. To model a system at different degrees of protonation (DPr), we chose the 20% DA CHS with half of the amino group protonated (DPr = 50%) to represent pH = 6.5, and further the same oligomer at DPr = 0% and DPr = 100%, meaning that the CHS chain is either fully deprotonated or fully protonated, corresponding to pH > 6.5 and pH < 4, respectively. Only the polymer charges were varied at the different pH. While the surface charge of the Mnt may also depend on pH, it has been shown that only the edge surfaces, i.e., (0 1 0) and (1 1 0), are affected whereas the adsorption plane (0 0 1) is nearly unaffected by the environmental pH.51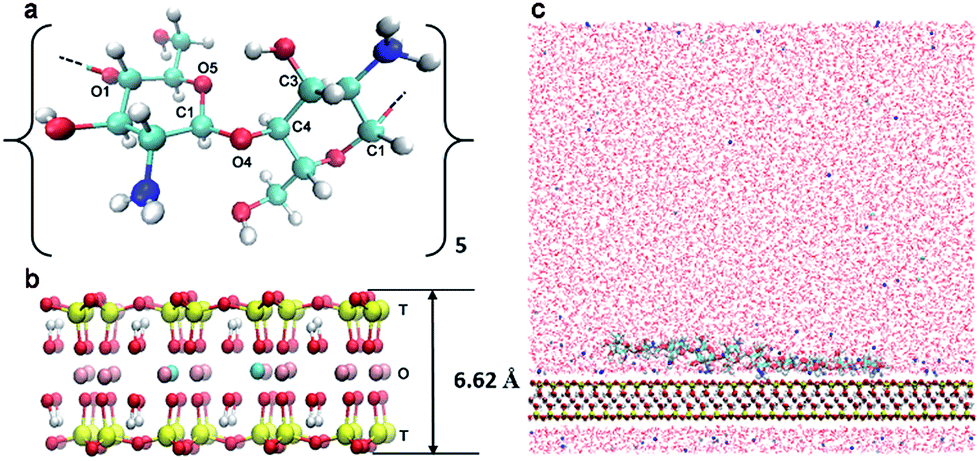 | ||
| Fig. 1 Molecular graphics representation of (a) molecular structure of one glucosamine dimer, the CHS or CHT chain formed by five dimers with different number of acetylated unit to present different DA; glucosidic linkage dihedral angles ϕ and ψ are defined by atoms O5 C1 O4 C4 and C1 O4 C4 C3, respectively. (b) Molecular structure of the super unit cell of Mnt clay: T, the tetrahedral layer; O, the octahedral layer. (c) The front view of the simulation box for CHS–Mnt or CHT–Mnt system. | ||
A summary of all the simulated chitosan and chitin models is shown in Table 1. The chains were all generated by the LEaP module26 from the AMBER suite of programs. The Mnt clay model was a sodium smectite was built based on the structure of pyrophyllite by Cygan et al.27 In the present study, the Mnt model was built from combining 8 super-cells in the x-direction, and 5 in the y-direction, with the chemical formula of each super cell being Na3[(Si31Al)](Al14Mg2)O80(OH)16 (Fig. 1b). The total negative charge was balanced by 120 Na+ ions.
| Systema | DAb (%) | DPrc (%) | pH | No. chain atomsd | No. ionse | No. waterf | Sequenceg |
|---|---|---|---|---|---|---|---|
| a CHS–Mnt: chitosan with montmorillonite clay, CHT–Mnt: chitin with montmorillonite clay; the number of Mnt atoms is 6400 and the total charge of Mnt clay in each system is −120. b Degree of acetylation (DA). c Degree of protonation (DPr). The three levels (0%, 50%, 100%) correspond to three pH levels, pH > 6.5, pH = 6.5, and pH < 4, respectively. d The number of atoms of the CHS or CHT chain. e The total number of sodium and chloride ions. f The total number of water molecules in the system. g The sequence of the CHS or CHT. g: glucosamine; p: protonated glucosamine; a: acetylated glucosamine. | |||||||
| CHS 0%–Mnt | 0 | 50 | pH = 6.5 | 228 | 125 | 17 431 | g-p-g-p-g-p-g-p-g-p |
| CHS 20%–Mnt | 20 | 50 | pH = 6.5 | 237 | 124 | 17 428 | g-p-a-g-p-g-p-a-g-p |
| CHS 20%–Mnt | 20 | 100 | pH < 4 | 241 | 128 | 17 358 | p-p-a-p-p-p-p-a-p-p |
| CHS 20%–Mnt | 20 | 0 | pH > 6.5 | 233 | 120 | 17 387 | g-g-a-g-g-g-g-a-g-g |
| CHS 40%–Mnt | 40 | 50 | pH = 6.5 | 246 | 123 | 17 328 | g-a-p-g-a-p-a-g-p-a |
| CHT 60%–Mnt | 60 | 50 | pH = 6.5 | 255 | 122 | 17 488 | g-a-p-a-a-g-a-a-p-a |
| CHT 80%–Mnt | 80 | 50 | pH = 6.5 | 264 | 121 | 17 426 | a-a-g-a-a-a-a-p-a-a |
| CHT 100%–Mnt | 100 | N/A | N/A | 273 | 120 | 17 416 | a-a-a-a-a-a-a-a-a-a |
The CHS–Mnt system thus consists of one oligosaccharide chain, and one Mnt sheet in aqueous solution (Fig. 1c) in a rectangular simulation box with a size of 8 × 9 × 8 nm3. Periodic boundary conditions (PBC) were applied in all three directions. Approximately 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 explicit water molecules were added as aqueous solution. Charges from CHS or CHT chains were balanced by adding chloride ions. The description of the simulated CHS–Mnt and CHT–Mnt systems are all summarized in Table 1.
400 explicit water molecules were added as aqueous solution. Charges from CHS or CHT chains were balanced by adding chloride ions. The description of the simulated CHS–Mnt and CHT–Mnt systems are all summarized in Table 1.
Computational details
All the calculations were performed by GROMACS/4.6.5 package.28–31 The GLYCAM_06h force field32,33 was applied to the acetylated glucosamine. For general glucosamine units and the protonated glucosamine unit, the partial atomic charges were derived by the ensemble average charge approach, the same method that was used in the original optimization of the GLYCAM_06 force field as well. Details of this derivation can be found in ESI.† The Mnt clay was modelled by the CLAYFF force field,27 and water was described by the simple point charge (SPC) water model.34 Non-bonded parameters for counter ions were taken from the work of Smith et al.35To build up the system, each oligomer was first solvated in a 5 × 5 × 5 nm3 box of water and equilibrated by a 3 ns long MD simulation with a linear annealing process from 0 K to 298 K. Following the solvent equilibration, a total of 120 ns further MD run with isothermal–isobaric (NPT) ensemble was performed to get well equilibrated conformations. Next, the chains were extracted and put on top of the Mnt surface with a center-of-mass distance between the oligomer and the clay of 1.2 nm. These systems were then subjected to 500 ps MD simulation under NVT conditions, followed by 1 ns in NPT. Finally the systems were solvated using explicit water. Each system was then subjected to a 200 ns long NPT production simulation, where the temperature was held constant at 298 K by coupling to the Nosé–Hoover thermostat,36,37 and the pressure was 1 bar in the direction of xx, yy and zz and 0 bar in the off-diagonal direction xy/yx, xz/zx, and yz/zy, by coupling to the Parrinello–Rahman barostat38,39. The compressibility was set to 10−6 bar−1 in all directions. The long-range electrostatic interactions were evaluated by the particle mesh Ewald (PME) method.40,41 Cut-off distance for the separation of direct and the reciprocal space was set to 1.2 nm. The non-bonded van der Waals forces were evaluated by a switching function, which started at 0.9 nm and reached zero at 1.0 nm. The all-bonds constraints were with the parallel version linear constraint solver (P-LINCS).42,43 The basic time step was 2 fs with the leapfrog algorithm44 and output data was saved every 2500 step.
To calculate the affinity of the oligosaccharide chains to the Mnt clays surface, steered molecular dynamic simulations (SMD) were carried out by pulling the single CHS or CHT chain away from the Mnt surface along the perpendicular direction (z axis). The free energy profile (corresponding to the work of adhesion WA), was computed as the potential of mean force (PMF) using umbrella sampling method,45,46 with the reaction coordinate being the z-component of the center-of-mass distance between the oligomer and the clay. A total of 150 umbrella windows were split from the SMD pulling trajectory and further subjected to 3 ns MD simulation each. The PMFs were finally generated from the last 2.5 ns of each umbrella window by the weighted histograms analysis method (WHAM)47 implemented in the GROMACS package. Each PMF is the average of three independent PMF calculations.
Results and discussion
Interfacial adhesion
It is well known that interfacial adhesion plays an important role for the mechanical behaviour of polymer–clay composite materials.17,20,25 The molecular adhesion between CHT/CHS oligomers and Mnt is here quantified as the molecular work of adhesion, WA, defined as free energy difference between a state where the oligomer is adsorbed and another state where it is completely detached. In the subsequent sections, the resulting adhesion as a function of acetylation and protonation respectively, is discussed and analyzed in terms of interaction between different components. To simplify the discussion, three types of functional groups in the CHT/CHS oligomers are identified and analyzed in detail. They are the acetyl group (chemical structure O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CH3, named Ac, residing on “a” units in the sequence in Table 1), the protonated amino group (named NH3+, residing on “p” units in Table 1) and the non-protonated amino group (named NH2, residing on “g” units in Table 1).
C–CH3, named Ac, residing on “a” units in the sequence in Table 1), the protonated amino group (named NH3+, residing on “p” units in Table 1) and the non-protonated amino group (named NH2, residing on “g” units in Table 1).
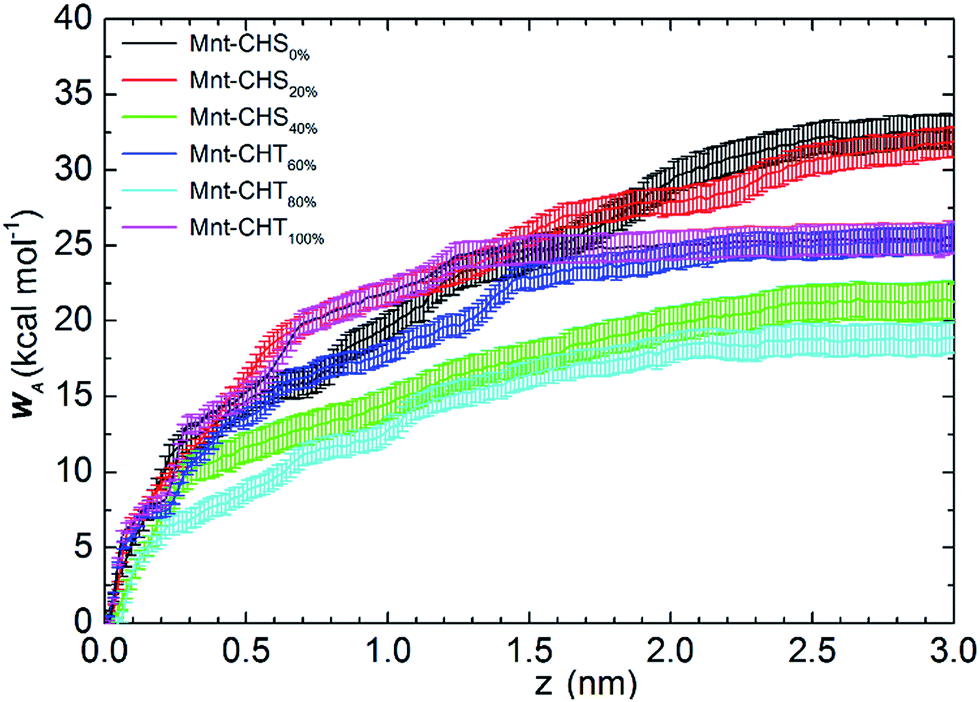 | ||
| Fig. 2 Free energy profiles for the studied CHS–Mnt or CHT–Mnt systems at different DA. The starting point of x-axis is normalized to the adsorbed conformation in each system; the distance (z) is measured along the perpendicular direction of the clay surface. The y-axis refers to the free energy difference, known as the work of adhesion (WA). | ||
When DA > 50%, and by definition has turned into CHT, the total WA remains comparable to that of the highly charged CHS. The same value, WA = 25.4 ± 1.0 kcal mol−1, is found for CHT at DA = 60% and 100%, while it is WA = 18.9 ± 1.0 kcal mol−1 for CHT at DA = 80%. Despite the oligomers gradually losing their charge, interaction with clay remains strong. When DA is above 50%, more than half of the sugar units in the polymer chain are acetylated, meaning that the functional group Ac is replacing one of the protons previously bound to the nitrogen atom of the (neutral) NH2 group, which is a process that also increases the molecular weight. Although the N-acetylated glucosamine sugar unit is neutral on the whole, the result of acetylation makes the total side group [Ac-NH] carry a negative partial charge (see Fig. S1 and Table S1 in ESI†). Since the Mnt surface is negatively charged as well, it is not likely the Ac groups interact directly with the clay, but there is, on the other hand, an opportunity to form strong electrostatic interactions with the positively charged Na+ ions.
To further test this hypothesis, we analyse the interfacial structure in terms of the density profiles of the different components for two extreme cases: CHS0%–Mnt and CHT100%–Mnt as shown in Fig. 3. Density profiles for all studied systems are provided in Fig. S2 in ESI.† From Fig. 3 it is obvious that the density of counter ions is always highest at the Mnt surface, where also the NH3+ nitrogens of CHS0% reside (Fig. 3 left). Next observation is that both NH2 nitrogens from CHS0% as well as the Ac oxygens from CHT100% are located in close proximity to the counter ions but not directly at the surface. In the case of Ac oxygens two distinct peaks are located near the peaks of counter ions and these are higher than the NH2 nitrogen peak in CHS0%–Mnt. We hypothesize that the strong adhesion between the highly acetylated but uncharged CHT100% and the Mnt is due to strong interaction between Ac and the counter ions that in turn are essentially glued to the surface. A look at the pictorial view of the interfaces as presented in Fig. 5 also supports the hypothesis.
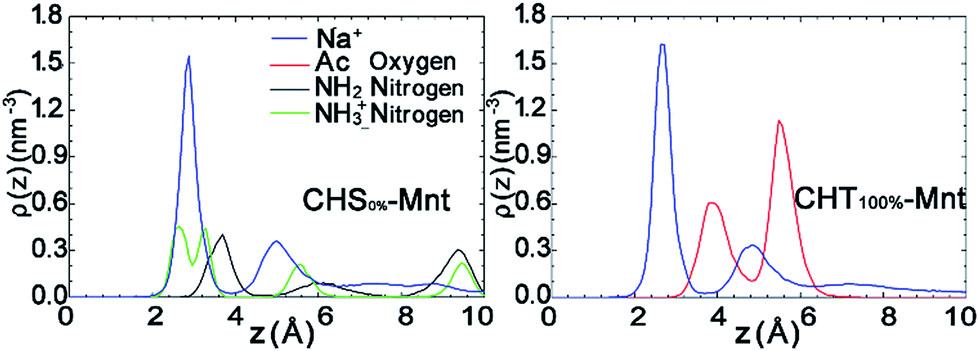 | ||
| Fig. 3 Density profiles of the different functional group atoms of the polymer and counter ions at the interfacial area of CHS0%–Mnt (left) and CHT100%–Mnt (right) systems. Density value of counter ions Na+ are divided by a factor of 5 to present all plots in a uniform range. Plots are calculated from the last 50 ns trajectories. | ||
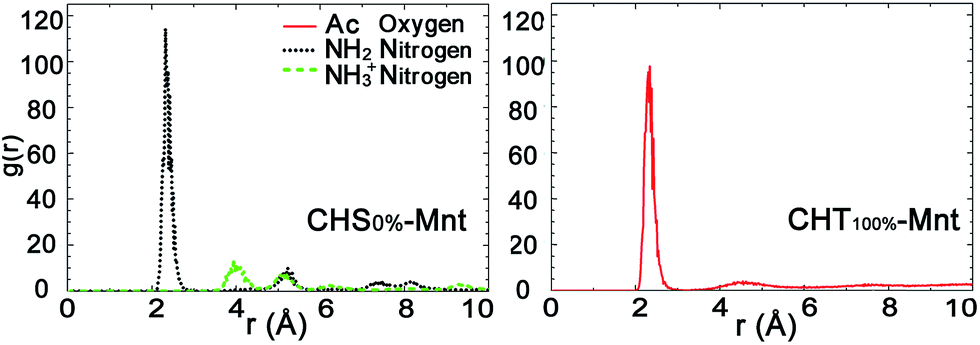 | ||
| Fig. 4 Radial distribution functions (RDFs) between the functional group atoms of the polymer and counterions Na+ in the CHS0%–Mnt (left) and CHT100%–Mnt (right) systems. Plots are calculated from the last 50 ns trajectories. | ||
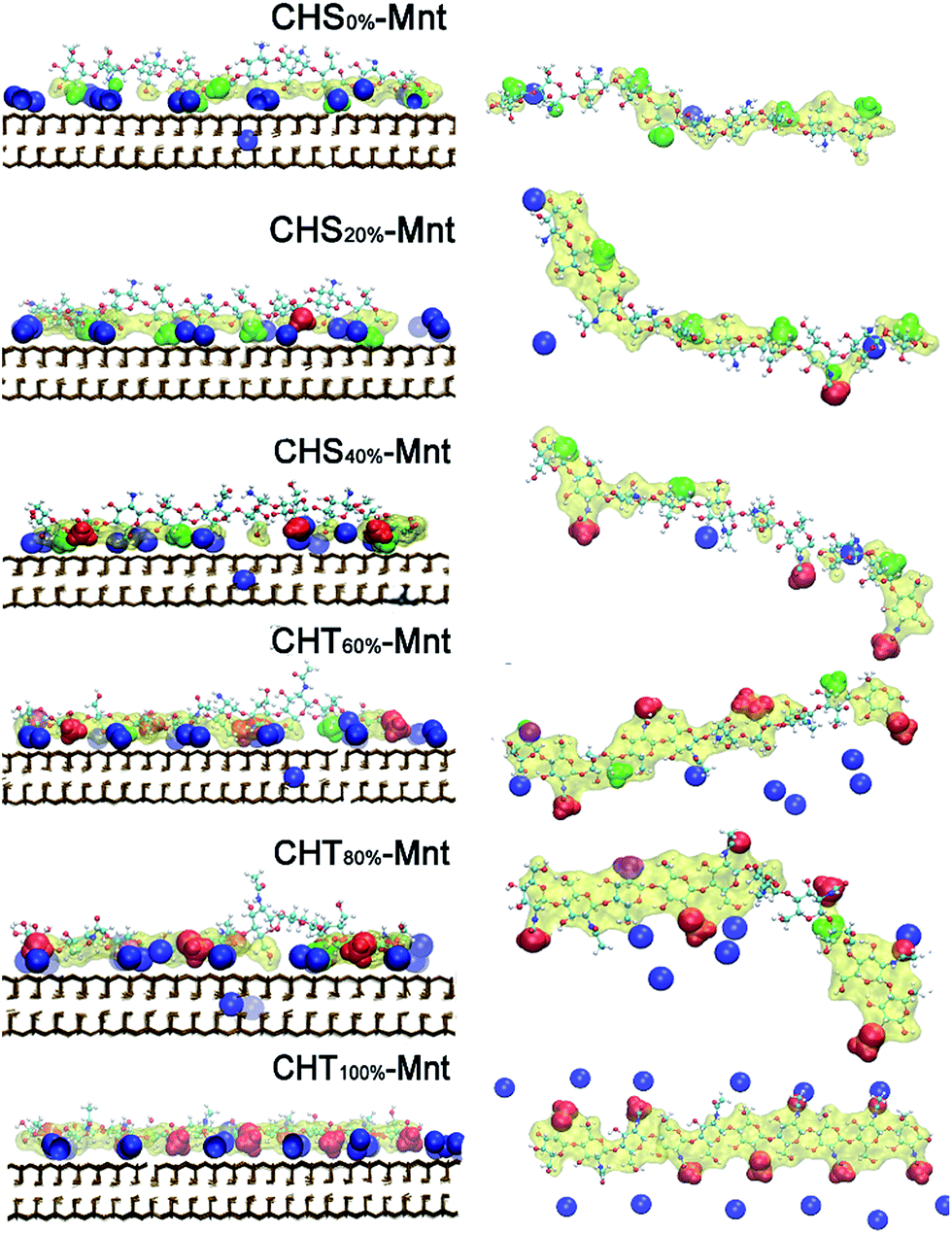 | ||
| Fig. 5 Snapshot of the equilibrated CHS or CHT–Mnt conformation. The left panel is in a front view corresponding to each CHS–Mnt or CHT–Mnt system at different DA. The right panel is in a top view of the conformation of CHS or CHT at different DA, corresponding to the left panel. Colour and drawing scheme: adsorbed Ac (OC–CH3): red; adsorbed NH2 group: magenta; adsorbed NH3+ group: green; adsorbed counter ions Na+: blue; adsorbed atoms from the CHS or CHT within 4 Å of Mnt clay surface: yellow transparent surface. | ||
Fig. 4 presents the radial distribution functions (RDFs) between the counter ions and each functional group atoms for the CHS0%–Mnt and CHT100%–Mnt systems. Other intermediate cases are shown in Fig. S3 in ESI.† The correlation between the NH2 nitrogen and sodium ions in CHS0%–Mnt is of similar strength as the correlation between Ac oxygen and sodium ions in CHT100%–Mnt. Since both atoms carry a substantial negative charge, this is understandable. In addition, we have calculated the dipole moment at the centre of mass for both functional groups. It was found to be 4.7 Debye for the Ac group, and 2.7 Debye for the NH2 group, indicating a difference that can partly explain why the contribution to the adhesion is more pronounced for the Ac group compared to the NH2 group. The NH3+ nitrogens are not coordinating the Na+ ions, since the total positive charge of the group repels the ions.
In summary, the molecular adhesion between CHS/CHT oligomers and Mnt is strong and comparable to the molecular adhesion found between xyloglucan and Mnt in K+ aqueous solution in our previous study (26.9 kcal mol−1).25 Two mechanisms are identified and assumed to affect the interaction between CHS/CHT and Mnt. First, when DA < 50%, electrostatic attraction between the positively charged CHS and the negatively charged Mnt surface is the main driving force for the polymer adsorption. Second, when DA > 50%, the increasing amount of Ac groups interact strongly with the sodium ions that practically act as an electrostatic “glue” between the polymer and the Mnt surface. Similar ion bridging effects have also been observed in other fields. For instance, in low salinity oil recovery,48 and biochemistry, where salt bridges are essential to protein stabilization.49
Free energy profiles of the CHS20% at different pH are shown in Fig. 6. As expected, the fully protonated (DPr = 100%) chitosan CHS20% exhibits the strongest adhesion to the Mnt with a value of 33.8 ± 1.0 kcal mol−1. The main reason is likely to be electrostatic attraction between the NH3+ protons and the negatively charged Mnt. Between high (DPr = 100%) and medium (DPr = 50%) protonation, a difference of only 2 kcal mol−1 is observed. However, the interaction is significantly weakened when the polymer is completely deprotonated (DPr = 0%) with a value of 8.56 ± 0.9 kcal mol−1 only. Hence, the effect of DPr is much stronger than the effect of DA in Fig. 2.
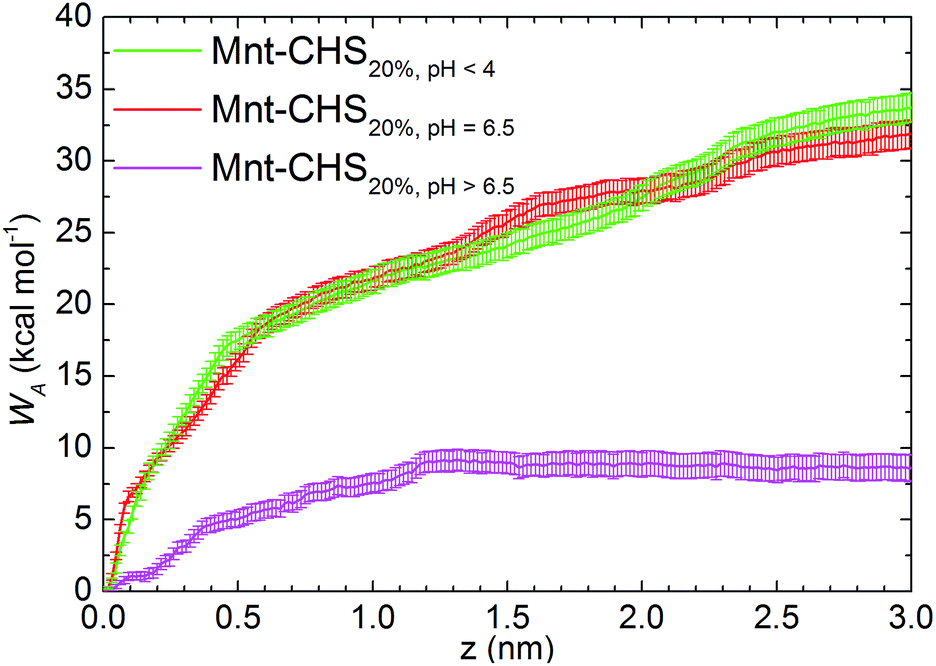 | ||
| Fig. 6 Free energy profiles for the CHS20%–Mnt systems at different DPr. The starting point of x-axis is normalized to the adsorbed conformation in each system; the distance (z) is measured along the perpendicular direction of the clay surface. The y-axis refers to the free energy difference, known as the work of adhesion WA. | ||
The density profiles in Fig. 7 in combination with the snapshot pictures in Fig. 8 support the hypothesis that the strong adsorption of CHS20% is due to the protonation in the acidic solution, where similarly to Fig. 3, the NH3+ nitrogens are found to be close to the surface. When pH is above 6.5 though, all amino groups are deprotonated and therefore no such attraction takes place. In this case, the NH2 nitrogens are found close to the Na+ ions, but apparently not experiencing the “glue” effect to the same extent as the Ac oxygens in CHT100% do in Fig. 2. Although both the N-acetylated and the non-acetylated glucosamine sugar units are electrically neutral, the functional group Ac carries less charge compared to the replaced proton, and hence the total side group [Ac-NH] turns more negative (−0.32e) than the NH2 group (−0.21e) (see Fig. S1 and Table S1 in ESI†), which can explain the observed difference.
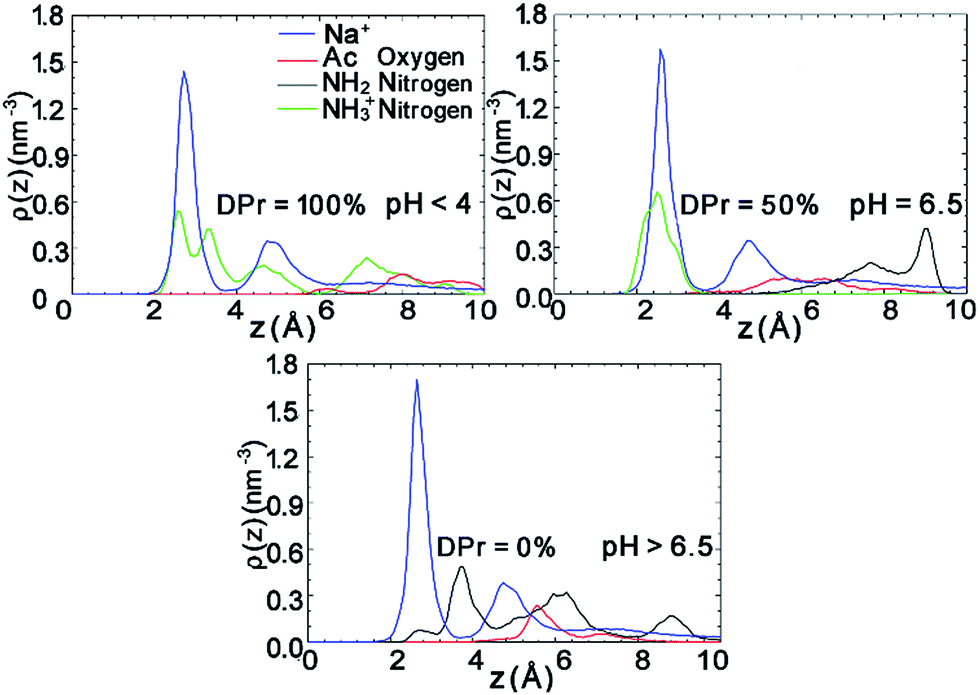 | ||
| Fig. 7 Density profiles of the different functional group atoms of the polymer and counter ions at the interfacial area of CHS20%–Mnt systems at different DPr. Density value of counter ions Na+ are divided by a factor of 5 to present all plots in a uniform range. Plots are calculated from the last 50 ns trajectories. | ||
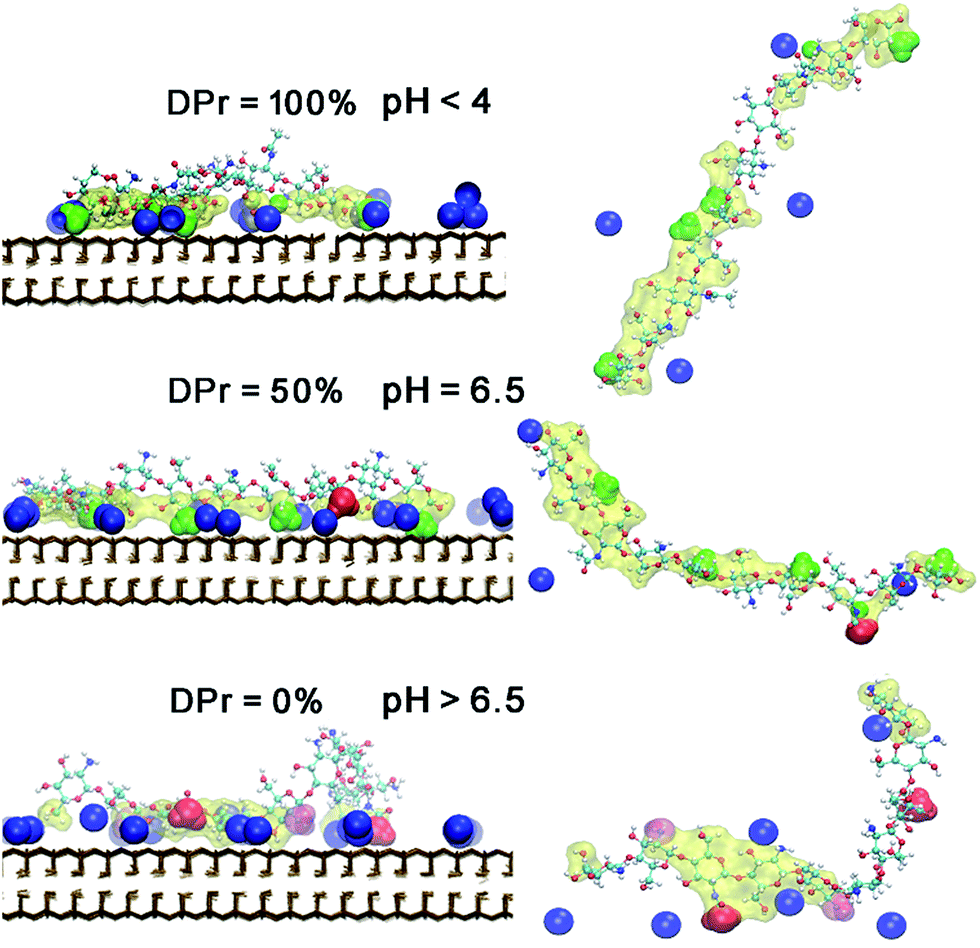 | ||
| Fig. 8 Snapshots of equilibrated CHS20%–Mnt complex at three different DPr. The left panel is in a front view. Right panel is in a top view of each CHS20% polymer, corresponding to the left panel. Colour and drawing scheme is the same as Fig. 5. | ||
Further analysis of the radial distribution functions in Fig. 9 indicate that the fully protonated polymer is not coordinating any Na+ ions at all, indicating direct interaction with the Mnt surface without the help of ions. The 50% protonated CHS has similar correlation to the Na+ ions from its two functional groups NH2 and Ac, indicating Na+ ions may play a role as a medium. Most importantly, for the fully deprotonated chitosan, by coordinating more Na+ ions, the functional Ac groups play a key role in the polymer–clay interaction for the same reason as we identified for the case of CHT100%. This is also seen in the snapshots in Fig. 8; there is no Ac group attached to the Mnt surface in the case of the fully protonated CHS, while one Ac group is found attached on the Mnt surface in the case of 50% protonated CHS. When CHS is completely deprotonated though, both available Ac groups are attached to the clay surface.
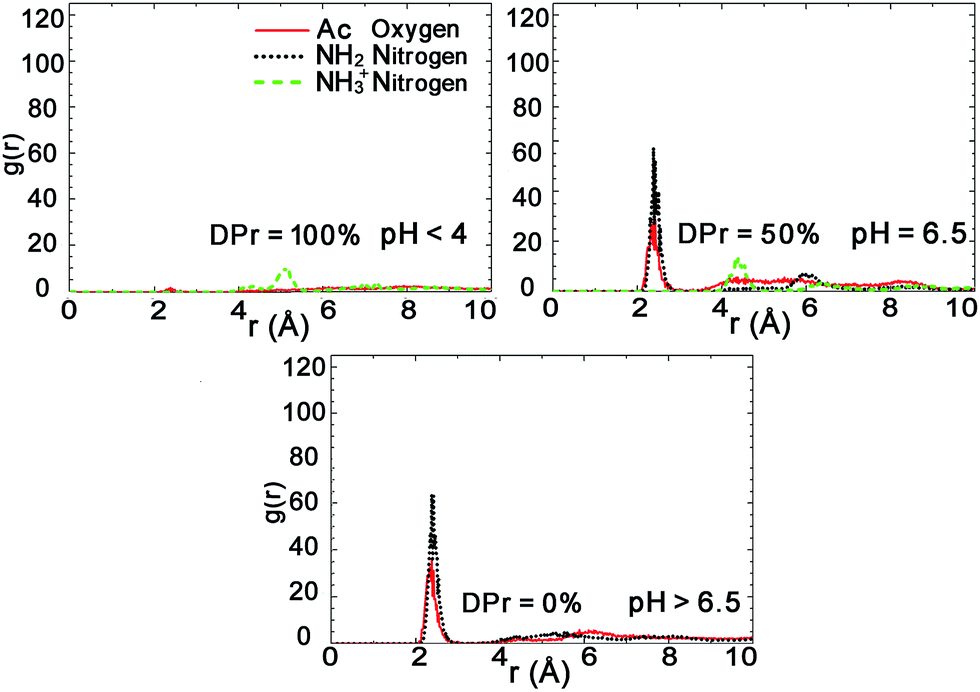 | ||
| Fig. 9 Radial distribution functions (RDFs) between the functional group atoms of CHS20% and counter ions Na+ for CHS20%–Mnt systems at different DPr. Plots are calculated from the last 50 ns trajectories. | ||
The role of hydrogen bonds
Just as intramolecular hydrogen bonds play an important role for the stabilization of the secondary structure in proteins, the extended two-fold helix of both chitin and chitosan is stabilized by the intramolecular hydrogen bond O3(n)–HO3(n)⋯O5(n+1) across the glycosidic linkage,52 which consequently play an important role for the chitin crystalline structure and directly contribute to the chain stiffness.8,9Errington and coworkers have reported that high acetylation makes the chitosan in solution adopt a coiled, but rigid conformation, whereas low acetylation gives a rod-like but flexible chain,53 which they attribute to that acetylation restricts the rotation around the glycosidic linkage. It thus seems that acetyl groups can contribute to the formation and stabilization of intramolecular hydrogen bonds.
Table 2 lists the number of intramolecular O3(n)–HO3(n)⋯O5(n+1) hydrogen bonds as a function of DA. Interestingly, for the adsorbed oligomers, the number of hydrogen bonds is severely affected by DA. As the number of acetyl groups increases, so does the number of hydrogen bonds. The effect on the structure can be clearly seen in Fig. 5, where the oligomers in the case of high DA adsorb in a flat extended conformation. For oligomers in solution, however, the number of intramolecular hydrogen bonds is constant for all DA's, within the error limits. The occupancy turns out to be around 50%, at any instant, regardless of DA. This is somewhat surprising as it stands in contrast to the findings of Errington et al.,53 but it might be that the comparison between short oligomers and large macromolecules is not completely fair when it comes to macroscopic conformational behavior.
| Polymer systems | Intramolecular hydrogen bonds | Polymer–water hydrogen bonds | |||||
|---|---|---|---|---|---|---|---|
| DA (%) | pH | O3(n)–HO3(n)⋯O5(n+1) | O3–HO3⋯OW | O3⋯HW–OW | |||
| Adsorbed form | Free in water | Adsorbed form | Free in water | Adsorbed form | Free in water | ||
| 0 | pH = 6.5 | 1.9 ± 0.7 | 4.9 ± 1.5 | 6.1 ± 1.2 | 3.1 ± 1.4 | 5.1 ± 1.4 | 7.8 ± 1.7 |
| 20 | pH < 4 | 1.7 ± 0.6 | 3.1 ± 1.4 | 5.5 ± 1.1 | 5.0 ± 1.5 | 3.2 ± 1.3 | 7.1 ± 1.7 |
| pH = 6.5 | 4.2 ± 0.9 | 4.9 ± 1.5 | 4.2 ± 1.1 | 3.3 ± 1.3 | 6.4 ± 1.5 | 8.3 ± 1.7 | |
| pH > 6.5 | 4.3 ± 1.4 | 6.6 ± 1.3 | 2.3 ± 1.4 | 2.2 ± 1.1 | 9.1 ± 1.8 | 10.3 ± 1.7 | |
| 40 | pH = 6.5 | 3.1 ± 0.9 | 4.7 ± 1.5 | 5.3 ± 0.9 | 3.4 ± 1.4 | 7.3 ± 1.5 | 8.9 ± 1.7 |
| 60 | pH = 6.5 | 2.1 ± 0.9 | 4.2 ± 1.3 | 2.1 ± 1.0 | 4.3 ± 1.3 | 8.0 ± 1.8 | 9.3 ± 1.8 |
| 80 | pH = 6.5 | 4.8 ± 0.9 | 5.1 ± 1.5 | 4.1 ± 0.9 | 3.5 ± 1.4 | 8.8 ± 1.7 | 10.5 ± 1.8 |
| 100 | N/A | 7.7 ± 1.0 | 5.4 ± 1.4 | 1.0 ± 0.8 | 3.3 ± 1.4 | 9.8 ± 1.8 | 11.3 ± 1.8 |
As a consequence of affecting the structure, the intramolecular hydrogen bonds also affect the accessibility of water, and thus the solvation of the oligosaccharide. More specifically, with the O3(n)–HO3(n)⋯O5(n+1) hydrogen bond intact, the hydroxyl oxygen O3 can only function as an acceptor for hydrogen bonds to water, whereas when the intramolecular hydrogen bond is broken, it can function as both acceptor and donor. From Table 2 we see that as DA increases, and the occupancy of the O3(n)–HO3(n)⋯O5(n+1) bond along with it, the number of O3–HO3⋯OW bonds decreases and the O3⋯HW bonds increases, accordingly. The total number of hydrogen bonds to water remains constant though.
Furthermore, the two situations have a significant impact on the orientation distribution of water in the vicinity of the O3–HO3 hydroxyl group. Following Franca et al.,8 we characterize the orientation of water molecules within a 0.5 nm radius from O3 as the cosine of the angle between the water dipole vector and the distance vector between O3 and the water oxygen (see Fig. 10). In this representation, a value of around −0.6 means that one of the water hydrogen atoms is pointing directly towards O3, which means that O3 in this case is functioning as an acceptor. A value close to 1 on the other hand indicates that O3 in that case is functioning as a donor. Fig. 10 plots this distribution, for all different DA. For high DA, there is a pronounced peak around −0.6 that gradually disappears as DA decreases, as expected. What is evident from Fig. 10 is that having O3 as acceptor leads to a much more localized orientation distribution, which clearly is disfavored by entropy and thus should be worse from a solvation perspective. A similar trend for the orientation distribution can be seen also for oligosaccharides in solution (see Fig. S4 in the ESI†), although considerably weaker, which correlates with the low impact DA has on hydrogen bonding in this case.
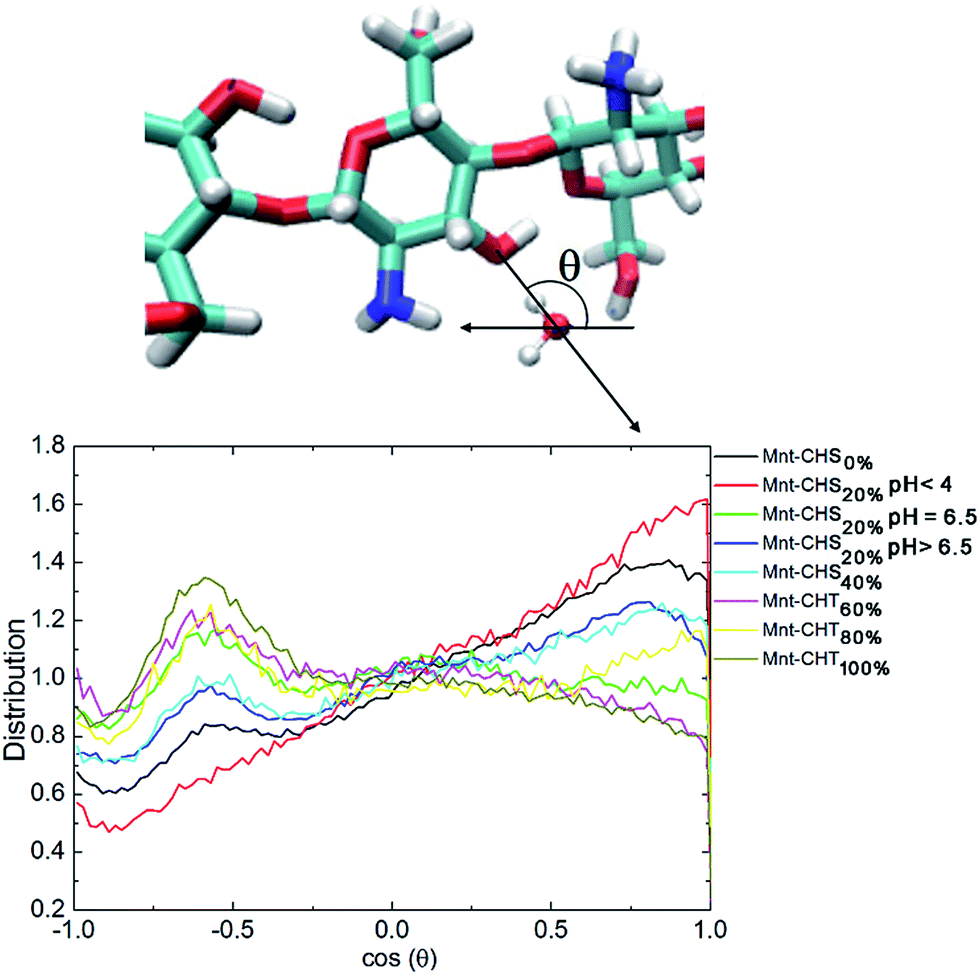 | ||
| Fig. 10 Orientation distribution of water molecules within a 0.5 nm radius of the hydroxyl O3 atom of the studied oligomers when adsorbed to Mnt. The horizontal axis shows the average value of the cosine of the angle θ, which is defined by two vectors: one is the water dipole vector, the other is the vector formed between the O3 and the water oxygen, as marked by the black arrows in the upper panel. Color scheme for the sugar unit in the upper panel: carbon (cyan), oxygen (red), hydrogen (white), nitrogen (blue). | ||
Taken altogether, hydrogen bonding, both intramolecular and to surrounding water molecules, is intimately linked to polysaccharide structure, and its solvation. For the adsorbed oligomers, there is clear correlation between the hydrogen bonding and the DA. However, given both that hydrogen bonds are highly opportunistic, and also the small variations with DA for the oligomers in solution, it is likely that the hydrogen bonding in the present case arises as a consequence of the imposed oligomer structure rather than the other way around.
Naturally, hydrogen bonds contribute to the overall energetics of the system, but as a driving force for adsorption, it is secondary to the mechanisms discussed earlier.
On a final note, the adhesion of the polymer to Mnt must clearly be affected by the polymer solubility.11,50 Thus, to investigate the thermodynamics of these systems in full, also the possibility for aggregation of polymers in solution will have to be brought into the equation. This would be an interesting topic for future work but is out of the scope of the present study.
Conclusions
In this study, we have investigated the adhesion of chitin and chitosan oligomers to Mnt clay in fully hydrated conditions. Specifically, the effects of N-acetylation and protonation on the work needed to detach the oligomers from the clay surface were studied using molecular dynamics simulations.Our calculations show surprisingly small variation in adhesion with DA. Despite the fact that the surface is negatively charged, and that the oligomers are positively charged at DA = 0%, +0.5e per sugar unit, and neutral at DA = 100%, the calculated free energies were 32.7 kcal mol−1 and 25.4 kcal mol−1, respectively, indicating strong adhesion in both cases.
Based on the distribution of the relevant functional groups (amino groups and acetyl groups) and counter ions close to the Mnt surface, we propose that this is due to two different mechanisms that work in concert. Positively charged amino groups are preferentially located as close to the Mnt surface as possible, which leads to strong electrostatic interaction with the negatively charged surface. Neutral acetyl groups on the other hand are positioned a bit further away from the surface, but are strongly correlated with the positive Na+ counter ions, which are in all cases stuck at the surface. Thus, in this case, counter ions seem to act as “glue” between the acetyl groups and the Mnt.
The amino groups are deprotonated at pH >6.5, which renders chitosan neutral at those conditions. Simulations of a neutral chitosan oligomer show that the adhesion to Mnt in this case is substantially lower than for the charged form, only 8.6 kcal mol−1, which also is much lower than for pure chitin. The analysis shows that the neutral amino groups are strongly correlated to the Na+ ions, just as the acetyl groups. The reason why the adhesion in this case is weaker can be traced to the respective charge distributions of the functional groups. Partial charges on the acetyl groups are generally larger than on the amino groups, leading to both higher total charge of the groups as a whole, and also larger dipole and higher multipole moments.
The two different mechanisms lead to different structures of the adsorbed molecules. Charged oligomers preferentially adsorb sideways, whereas neutral oligomers adsorb flat down, which has consequences for the hydrogen bonding characteristics. The occupancy of the intramolecular O3(n)–HO3(n)⋯O5(n+1) hydrogen bond increases with DA, leading to more extended and less flexible conformations. At low DA, the more open structure makes the oligomers more accessible to solvation by surrounding water.
In summary, our calculations present new insights of how acetylation and protonation affect the adhesion of chitosan and chitin to charged surfaces, e.g. Mnt clay. This work is of potential interest for the designing of chitosan–Mnt composite materials. Furthermore, as a direct consequence of our proposed mechanism for the interaction between chitin/chitosan and Mnt, we predict that there is an effect from using different counter ions in the systems, and also that this effect should get stronger as the DA increases. In addition, setting polymer solubility issues aside, the moisture sensitivity of the final composite should be mildly dependent on both DA and the pH during processing.
Acknowledgements
Computational resources are supported by a grant from the Swedish National Infrastructure for Computing (SNIC) through the projects “Modeling of molecular interaction between nanoparticles and polymeric molecules” (SNIC 2013-26-31) and “Molecular Dynamics simulations of the interactions of organic polymers and inorganic clays” (SNIC 001-12-203). Computational support from PDC Centre for High Performance Computing (PDC-HPC) in KTH is acknowledged. Molecular graphics were produced using VMD.54 Funding from SSF FireFoam (author JW), SSF (ICA10-0086, author MB-W) and China Scholarship Council (author YW) is gratefully acknowledged.References
- A. Okada and A. Usuki, Macromol. Mater. Eng., 2006, 291, 1449–1476 CrossRef CAS.
- E. Ruiz-Hitzky, P. Aranda, M. Darder and G. Rytwo, J. Mater. Chem., 2010, 20, 9306–9321 RSC.
- J. L. Suter, R. L. Anderson, H. C. Greenwell and P. V. Coveney, J. Mater. Chem., 2009, 19, 2482–2493 RSC.
- B. K. G. Theng, Clays Clay Miner., 1982, 30, 1–10 CAS.
- D. W. Lee, H. Lim, H. N. Chong and W. S. Shim, Open Biomater. J., 2009, 1, 10–20 CrossRef CAS.
- G. Laufer, C. Kirkland, A. A. Cain and J. C. Grunlan, ACS Appl. Mater. Interfaces, 2012, 4, 1643–1649 CAS.
- K. Rudall and W. Kenchington, Biol. Rev., 1973, 48, 597–633 CrossRef CAS.
- E. F. Franca, L. C. Freitas and R. D. Lins, Biopolymers, 2011, 95, 448–460 CrossRef CAS PubMed.
- E. F. Franca, R. D. Lins, L. C. G. Freitas and T. P. Straatsma, J. Chem. Theory Comput., 2008, 4, 2141–2149 CrossRef CAS PubMed.
- A. Tiraferri, P. Maroni, D. Caro Rodríguez and M. Borkovec, Langmuir, 2014, 30, 4980–4988 CrossRef CAS PubMed.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- M. Darder, M. Colilla and E. Ruiz-Hitzky, Chem. Mater., 2003, 15, 3774–3780 CrossRef CAS.
- J. Wang, Q. Cheng and Z. Tang, Chem. Soc. Rev., 2012, 41, 1111–1129 RSC.
- Z. Tang, N. A. Kotov, S. Magonov and B. Ozturk, Nat. Mater., 2003, 2, 413–418 CrossRef CAS PubMed.
- A. Walther, I. Bjurhager, J.-M. Malho, J. Pere, J. Ruokolainen, L. A. Berglund and O. Ikkala, Nano Lett., 2010, 10, 2742–2748 CrossRef CAS PubMed.
- A. Walther, I. Bjurhager, J.-M. Malho, J. Ruokolainen, L. Berglund and O. Ikkala, Angew. Chem., Int. Ed., 2010, 49, 6448–6453 CrossRef CAS PubMed.
- J. J. Kochumalayil, M. Bergenstråhle-Wohlert, S. Utsel, L. Wågberg, Q. Zhou and L. A. Berglund, Biomacromolecules, 2013, 14, 84–91 CrossRef CAS PubMed.
- J. J. Kochumalayil, S. Morimune, T. Nishino, O. Ikkala, A. Walther and L. A. Berglund, Biomacromolecules, 2013, 14, 3842–3849 CrossRef CAS PubMed.
- H. B. Yao, Z. H. Tan, H. Y. Fang and S. H. Yu, Angew. Chem., Int. Ed., 2010, 49, 10127–10131 CrossRef CAS PubMed.
- P. Podsiadlo, Z. Tang, B. S. Shim and N. A. Kotov, Nano Lett., 2007, 7, 1224–1231 CrossRef CAS PubMed.
- R. A. Cunha, E. F. Franca, T. A. Soares, V. H. Rusu, F. J. S. Pontes and R. D. Lins, The Molecular Structure and Conformational Dynamics of Chitosan Polymers: An Integrated Perspective from Experiments and Computational Simulations, 2012 Search PubMed.
- H. Heinz, R. A. Vaia, R. Krishnamoorti and B. L. Farmer, Chem. Mater., 2007, 19, 59–68 CrossRef CAS.
- J. L. Suter and P. V. Coveney, Soft Matter, 2009, 5, 2239–2251 RSC.
- Y. Wang, J. Wohlert, L. A. Berglund, Y. Tu and H. Ågren, J. Mater. Chem. A, 2014, 2, 9541–9547 CAS.
- Y. Wang, J. Wohlert, M. Bergenstråhle-Wohlert, J. J. Kochumalayil, L. A. Berglund, Y. Tu and H. Ågren, Biomacromolecules, 2015, 16, 257–265 CrossRef CAS PubMed.
- T. J. Macke, W. Svrcek-Seiler, R. A. Brown, I. Kolossváry, Y. J. Bomble, D. A. Case, W. Zhang, T. Hou, C. Schafmeister and W. S. Ross, AmberTools Users’ Manual, 2010 Search PubMed.
- R. T. Cygan, J. J. Liang and A. G. Kalinichev, J. Phys. Chem. B, 2004, 108, 1255–1266 CrossRef CAS.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- D. van der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1719 CrossRef CAS PubMed.
- E. Lindahl, B. Hess and D. van der Spoel, J. Mol. Model., 2001, 7, 306–317 CAS.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- M. B. Tessier, M. L. DeMarco, A. B. Yongye and R. J. Woods, Mol. Simul., 2008, 34, 349–364 CrossRef CAS PubMed.
- K. N. Kirschner, A. B. Yongye, S. M. Tschampel, J. González-Outeiriño, C. R. Daniels, B. L. Foley and R. J. Woods, J. Comput. Chem., 2008, 29, 622–655 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren and J. Hermans, Interaction models for water in relation to protein hydration, Springer, Netherlands, 1981 Search PubMed.
- D. E. Smith and L. X. Dang, J. Chem. Phys., 1994, 100, 3757–3766 CrossRef CAS.
- S. Nosé, Mol. Phys., 1984, 52, 255–268 CrossRef.
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef.
- S. Nosé and M. L. Klein, Mol. Phys., 1983, 50, 1055–1076 CrossRef.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- B. Hess, J. Chem. Theory Comput., 2007, 4, 116–122 CrossRef PubMed.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- R. W. Hockney, S. P. Goel and J. W. Eastwood, J. Comput. Phys., 1974, 14, 148–158 CrossRef.
- G. M. Torrie and J. P. Valleau, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- J. Kästner, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 932–942 CrossRef.
- J. S. Hub, B. L. de Groot and D. van der Spoel, J. Chem. Theory Comput., 2010, 6, 3713–3720 CrossRef CAS.
- R. A. Nasralla and H. A. Nasr-El-Din, J. Pet. Sci. Eng., 2014, 122, 384–395 CrossRef CAS.
- M. Schledorn, B. H. Meier and A. Böckmann, Front. Mol. Biosci., 2015, 2, 14 Search PubMed.
- P. K. Dutta, J. Dutta and V. Tripathi, J. Sci. Ind. Res., 2004, 63, 20–31 CAS.
- X. Liu, X. Lu, J. Cheng, M. Sprik and R. Wang, Geochim. Cosmochim. Acta, 2015, 160, 91–99 CrossRef CAS.
- K. Ogawa, T. Yui and K. Okuyama, Int. J. Biol. Macromol., 2004, 34, 1–8 CrossRef CAS PubMed.
- N. Errington, S. Harding, K. Vårum and L. Illum, Int. J. Biol. Macromol., 1993, 15, 113–117 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the parameterization of protonated and deprotonated glucosamine; charge distributions of atoms in glucosamine, protonated glucosamine and acetylated glucosamine units; density profiles of the different functional group atoms for all studied CHS or CHT combined Mnt clay systems; radial distribution functions between the functional group atoms and counter ions for all the simulated systems; interaction energies between polymer and clay, and between polymer and water; and water orientations around the hydroxyl O3 atom of the CHS and CHT chains in solution. See DOI: 10.1039/c5ra06424d |
| This journal is © The Royal Society of Chemistry 2015 |