
Influence of electron acceptors on the kinetics of metoprolol photocatalytic degradation in TiO2 suspension. A combined experimental and theoretical study†
S. J.
Armaković
a,
S.
Armaković
b,
N. L.
Finčur
a,
F.
Šibul
a,
D.
Vione
c,
J. P.
Šetrajčić
b and
B. F.
Abramović
*a
aDepartment of Chemistry, Biochemistry and Environmental Protection, Faculty of Sciences, University of Novi Sad, Trg Dositeja Obradovića 3, 21000 Novi Sad, Serbia. E-mail: biljana.abramovic@dh.uns.ac.rs; Fax: +381 21454065; Tel: +381 214852753
bDepartment of Physics, Faculty of Sciences, University of Novi Sad, Trg Dositeja Obradovića 4, 21000 Novi Sad, Serbia
cDipartimento di Chimica, Università di Torino, Via Pietro Giuria 5, 10125 Torino, Italy
First published on 8th June 2015
Abstract
Metoprolol (MET) belongs to a group of frequently used β1-blockers, which often occur in waste waters. The objective of this work was to employ liquid chromatography (LC) and total organic carbon methods to study the photocatalytic degradation of MET in UV irradiated aqueous suspensions of TiO2 (Wackherr's “Oxyde de titane standard” and Degussa P25), in the presence of different electron acceptors such as molecular oxygen, hydrogen peroxide, potassium bromate, and ammonium persulfate. The degradation rates were found to be strongly influenced by the kind of electron acceptor and the type of catalyst. The optimal amount of hydrogen peroxide and potassium bromate was investigated as well. MET photocatalytic degradation was the fastest in the presence of O2 and potassium bromate with TiO2 Degussa P25, while mineralization was most efficient in the presence of molecular oxygen alone. In all investigated cases, degradation followed a pseudo-first order kinetics. Reaction intermediates of MET degradation in the presence of different electron acceptors with both catalysts were studied in detail and a number of them were indentified using LC-ESI-MS/MS. The interactions with MET of reactive radical species relevant to this study (O2˙−, ˙OH, BrO2˙, and SO4˙−) were theoretically investigated by means of density functional theory (DFT) computations.
1 Introduction
Studies dating to more than 30 years ago dealt with photocatalytic reactions, which erupted into the scientific literature when they were proposed as a suitable tool to promote separation of molecular H2 and molecular O2 from water using solar irradiation.1,2 Heterogeneous photocatalysis using TiO2 powders has become a subject of increasing interest during the past twenty years, mainly in the field of environmental protection and wastewater decontamination.3 Photocatalysis in the presence of semiconductors is triggered by the interaction of electrons and holes, generated in a photochemically activated solid, with the surrounding medium. Activation is the consequence of light absorption: the irradiation of the photocatalytic material with sufficient energy leads to the formation of holes (h+) in the valence band and electrons (e−) in the conduction band.The two species can either recombine or participate in reductive and oxidative reactions that lead to the decomposition of contaminants.4
The h+ can either directly oxidize pollutants, or oxidize water (preferably the OH− groups adsorbed on the solid surface) to produce ˙OH. In contrast, e− reduces surface-adsorbed O2. The oxidative and reductive reaction steps taking place with the irradiated photocatalyst (TiO2) are expressed as follows:5–8
| TiO2 + hν → e− + h+ | (1) |
| h+ + organic compound → CO2 + H2O + inorganic ions | (2) |
| H2O + h+ → ˙OH + H+ | (3) |
| O2 + e− → O2˙− | (4) |
A practical problem in using TiO2 as a photocatalyst is the energy waste due to the e−–h+ recombination, which results in lower degradation efficiency. The prevention of recombination is thus very important, and it can be achieved by adding proper electron acceptors. In the simplest systems, in aerated solution, molecular O2 acts as electron acceptor to prevent e−–h+ recombination.9 Additional oxidants, such as H2O2, S2O82−, and BrO3−, can act as electron acceptors to enhance the photodegradation efficiency. These electron acceptors can have several effects including: (I) avoidance of e−–h+ recombination because of scavenging of conduction-band electrons; (II) increase of the concentration of ˙OH and (III) production of other oxidizing species that can enhance the oxidation rate of the substrate and of its intermediate compounds.10
By employing computer simulations within the framework of the density functional theory (DFT), it is possible to gain insight into the changes of the investigated structures as a consequence of the presence of other molecules in the system.11,12 The information thus obtained is very important to further understand the degradation mechanisms of the investigated compounds.13,14
Fukui functions and Fukui indices are often used as local quantum-molecular descriptors. Fukui functions describe the changes in the molecular electron density as a consequence of the addition or removal of charge, while Fukui indices represent scalar values for each atom. Fukui functions are visualized as iso-surfaces, and larger Fukui values indicate higher reactivity.15,16 It should be emphasized that values of Fukui functions are sensitive to changes in basis sets and population analysis. Therefore, one shouldn't use these values as absolute but rather as comparative parameters.
The molecular electrostatic potential (MEP) is related to the charge distribution and it is a very useful descriptor to determine potential sites prone to electrophilic attack and nucleophilic reactions.14,17,18 In the field of pollutant degradation, MEP enables the localization of parts of a molecule that are prone to various types of attacks, also giving information on how molecules interact with other molecules or radicals. The MEP (hereafter indicated as V(r)), when neglecting polarization and nuclear rearrangement effects due to the presence of a unit test charge at the distance r, is given as follows:
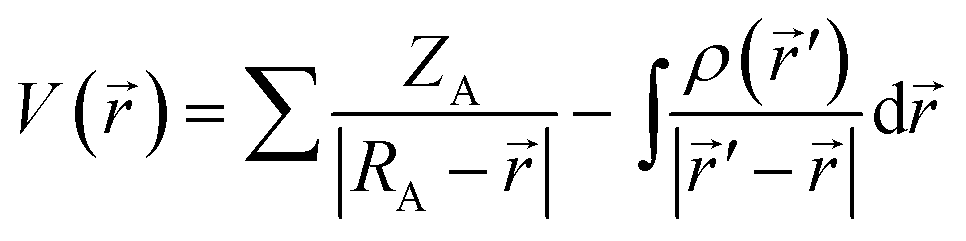 | (5) |
Natural bond order (NBO) can be used for efficient investigation of intra- and inter-molecular bonding and interactions. It is a convenient basis for the investigation of charge transfer or conjugative interactions in molecular systems.21–23 NBO analysis is carried out by examining all possible interactions between ‘filled’ (donor) NBOs and ‘empty’ (acceptor) NBOs, estimating their energetic importance by 2nd-order perturbation theory. In this way one obtains the energies of delocalization of electrons from filled NBOs into empty NBOs, e.g. stabilization energies gained by donation from the donor NBO to the acceptor NBO. For each donor NBO (i) and acceptor NBO (j), the stabilization energy associated with i → j delocalization can be estimated on the basis of the second-order perturbation theory:24–26
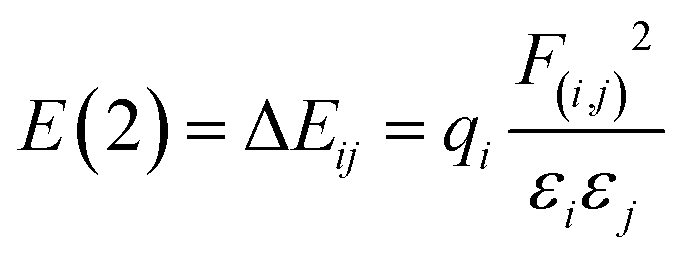 | (6) |
Metoprolol (MET) is a selective β1-blocker of the cardiac adrenergic receptors.27 Due to the frequent use, MET is present in sewage waters, in rivers from Netherlands (25–100 ng L−1),28 Poland (51–155 ng L−1),29 UK (7–11 ng L−1),30 Sweden (60–70 ng L−1),30 and Germany (exceeding 1000 ng L−1).31
Previous work has shown that the rate of MET photocatalytic degradation tended to a plateau at about 0.5–1.0 mM initial concentration of the substrate, and that photodegradation was most efficient at a photocatalyst loading of 1.0 mg mL−1.13 The aim of this work was to compare the kinetics of photodegradation of MET, sensitized by TiO2 Wackherr or Degussa P25 in aqueous suspension under the same experimental conditions, in the presence of different electron acceptors (O2, H2O2, S2O82−, and BrO3−). To monitor MET removal and mineralization, liquid chromatography (LC) and total organic carbon (TOC) analysis were used, respectively. An attempt has also been made to identify the intermediates formed during the photooxidation of MET in the presence of different electron acceptors with both catalysts, and a number of them were indentified using LC-ESI-MS/MS. Employing DFT computations, the interactions of radical species (O2˙−, ˙OH, BrO2˙, and SO4˙−) with MET and their possible effects on its degradation were investigated from the aspect of structural considerations, charge distribution, NBO analysis, Fukui functions and Fukui indices.
2 Materials and methods
2.1 Chemicals and solutions
All chemicals were of reagent grade and were used without further purification. The drug (±)-metoprolol(+)-tartrate salt (Sigma-Aldrich) was used as received (≥99% purity); 85% H3PO4 was purchased from Lachema, Neratovice; acetonitrile (ACN) was a product of J. T. Baker. Other chemicals were as follows: 30% H2O2 from Sigma-Aldrich; KBrO3 and (NH4)2S2O8 from Merck. All solutions were made using doubly distilled water. The used catalysts were TiO2 Degussa P25 (75% anatase and 25% rutile, surface area 50 ± 1.0 m2 g−1, crystallite size about 20 nm, non-porous) and TiO2 Wackherr's “Oxyde de titane standard” (100% anatase, surface area 8.5 ± 1.0 m2 g−1, crystallite size 300 nm, hereafter “TiO2 Wackherr”), produced by the sulfate process.322.2 Photodegradation procedures
Photocatalytic degradation was carried out in a cell made of Pyrex glass (total volume of ca. 40 mL, liquid layer thickness 35 mm), with a plain window on which the light beam was focused. The cell was equipped with a magnetic stirring bar and a water circulating jacket. A 125 W high-pressure mercury lamp (Philips, HPL-N, emission bands in the UV region at 304, 314, 335 and 365 nm, with maximum emission at 365 nm) together with an appropriate concave mirror was used as the radiation source. The output of the mercury lamp was calculated to be ca. 8.8 × 10−9 einstein mL−1 min−1 (potassium ferrioxalate actinometry).Experiments were performed using 20 mL of 0.05 mM MET containing 1.0 mg mL−1 of TiO2 (Wackherr or Degussa P25), except for the study of direct photolysis. The aqueous suspension of TiO2 was sonicated (50 Hz) in the dark for 15 min before irradiation, in order to uniformly disperse the photocatalyst particles and to attain adsorption equilibrium. Before irradiation, the suspension thus obtained was thermostated at 25 ± 0.5 °C in a stream of O2 (3.0 mL min−1), except for a control run in the absence of electron acceptors when N2 was bubbled (3.0 mL min−1) to remove dissolved oxygen. During irradiation, the mixture was stirred at a constant rate under continuous gas flow. All experiments were performed at the natural pH which changed during the photodegradation, from pH 7 to pH 4 in the case of TiO2 Wackherr and from pH 7 to pH 5 in the case of Degussa P25. In the investigation of the influence of electron acceptors, apart from constant O2 bubbling, solutions of H2O2, KBrO3 or (NH4)2S2O8 (at typical 3.0 mM initial concentration) were added to the MET solution.
2.3 Analytical procedures
The photodegradation of MET was monitored by liquid chromatography-photodiode array detection (LC-PDA). To do so, aliquots of 0.30 mL were taken from the reaction mixture at the beginning of the experiment and at regular time intervals. Aliquot sampling caused a maximum volume variation of ca. 10% in the reaction mixture. The suspensions were filtered through Millipore (Millex-GV, 0.22 μm) membrane filters to eliminate the photocatalyst. Lack of adsorption of MET on the filters was preliminarily checked. Afterwards, a 10 μL sample was injected and analyzed using a Shimadzu UFLC-PDA, equipped with an Eclipse XDB-C18 column (150 mm × 4.6 mm i.d., particle size 5 μm, 25 °C). The UV/vis PDA detector was set at 225 nm (wavelength of MET maximum absorption), as well as at 210, 260, 270 and 280 nm for the monitoring of the intermediates. The mobile phase (flow rate 0.8 mL min−1) was a mixture of ACN and water (the latter acidified with 0.1% H3PO4), with the following gradient: 15% ACN at 0 min, which was increased to 30% ACN in 5 min, after which 30% ACN was constant for 5 min; post time was 3 min. The retention time for MET was 6.0 ± 0.1 min. Reproducibility of repeated runs was around 3–10%.Concerning TOC analysis, 10 mL aliquots of the reaction mixture were taken at regular time intervals, diluted to 25 mL and analyzed after filtration on an Elementar Liqui TOC II analyzer, according to Standard US 120 EPA Method 9060A. For the LC-ESI-MS/MS evaluation of intermediates after 10 min irradiations, 100 μL samples were analyzed on an Agilent Technologies 1200 series LC with Agilent Technologies 6410A series electrospray ionization triple-quadrupole MS/MS, using Agilent Technologies Zorbax XDB-C18 column (50 × 4.6 mm i.d., particle size 1.8 μm, 40 °C). The mobile phase (flow rate 0.5 mL min−1) consisted of 0.05% aqueous formic acid and MeOH (gradient, 0 min 20% MeOH, 10 min 60% MeOH, 12 min 100% MeOH, post-time 3 min). Analytes were ionized using the electrospray ion source, a capillary voltage of 4.0 kV and with nitrogen as the drying gas (temperature 350 °C, flow 10 L min−1) and nebulizer gas (45 psi). High-purity nitrogen was used as the collision gas. Full scan mode (m/z range 50–800, scan time 100 ms, fragmentor voltage 100 V) in positive ion mode was used to select precursor ions for the starting compound and each degradation intermediate, as well as to examine isotopic peak distribution. Then, the product ion scan MS2 mode (fragmentor voltage 135 V, scan time 200 ms, collision energy 10–40 V in increments of 10 V) was used to elucidate the structure of each degradation intermediate.
2.4 Computational details
All DFT calculations were carried out using the Gaussian 03 software package,33 except for Fukui functions and Fukui indices that were calculated at the same level of theory using Jaguar, version 8.4,34 as implemented in the Schrödinger Materials Suite, release 2014-2. For the purpose of NBO analysis, it was used the NBO 3.0 program as implemented in Gaussian 03. For all systems, calculations were performed employing the B3LYP exchange and correlation functional with 6-31 G+(d) basis set.35 Two stages took place for all configurations. Firstly, equilibrium geometry of the investigated systems was located using default convergence criteria and, secondly, the harmonic vibrational spectrum was checked to assure that the true minimum of potential energy, characterized by positive frequencies, was located.In this work we investigated five systems: MET, MET/O2˙−, MET/O2˙−/˙OH, MET/O2˙−/BrO2˙, and MET/O2˙−/SO4˙−. To make the simulations more realistic, solvent effects of water were taken into account using the default Polarizable Continuum Model (PCM). Initially, in all cases O2˙− was placed above the hydroxyl group located at the tail of MET, while ˙OH, BrO2˙ and SO4˙− were placed above the aromatic ring.
Interesting sites containing a significant amount of charges were located through MEP surfaces, which were obtained using Molekel version 5.4 after geometry optimization and frequency check.36
3 Results and discussion
3.1 Effect of electron acceptors
Apart from O2, which is the most frequently applied electron acceptor, in this work we also investigated the influence of H2O2, BrO3−, and S2O82−. These compounds operate as e− scavengers and should be able to prevent e−–h+ recombination, thereby enhancing the formation of ˙OH and other reactive species.5,6 However, compounds such as H2O2 are also able to scavenge the photogenerated transients.37Fig. 1 and Table 1 show the time trend and degradation kinetics of MET, upon irradiation in the presence of TiO2 (Wackherr, 1a, and Degussa P25, 1b) and several electron acceptors. In all investigated cases, the degradation process followed a pseudo-first order kinetics. Note that the role of O2 as electron acceptor was investigated by comparing the system in air (blue triangles) with the one in which oxygen was bubbled to create an O2 atmosphere (purple stars; in such a system, the concentration of both gas-phase and dissolved O2 is expected to be ∼5 times higher than in the case of air equilibrium). For further comparison, the behaviour of the deoxygenated system (N2 bubbling, pink triangles) was also studied.
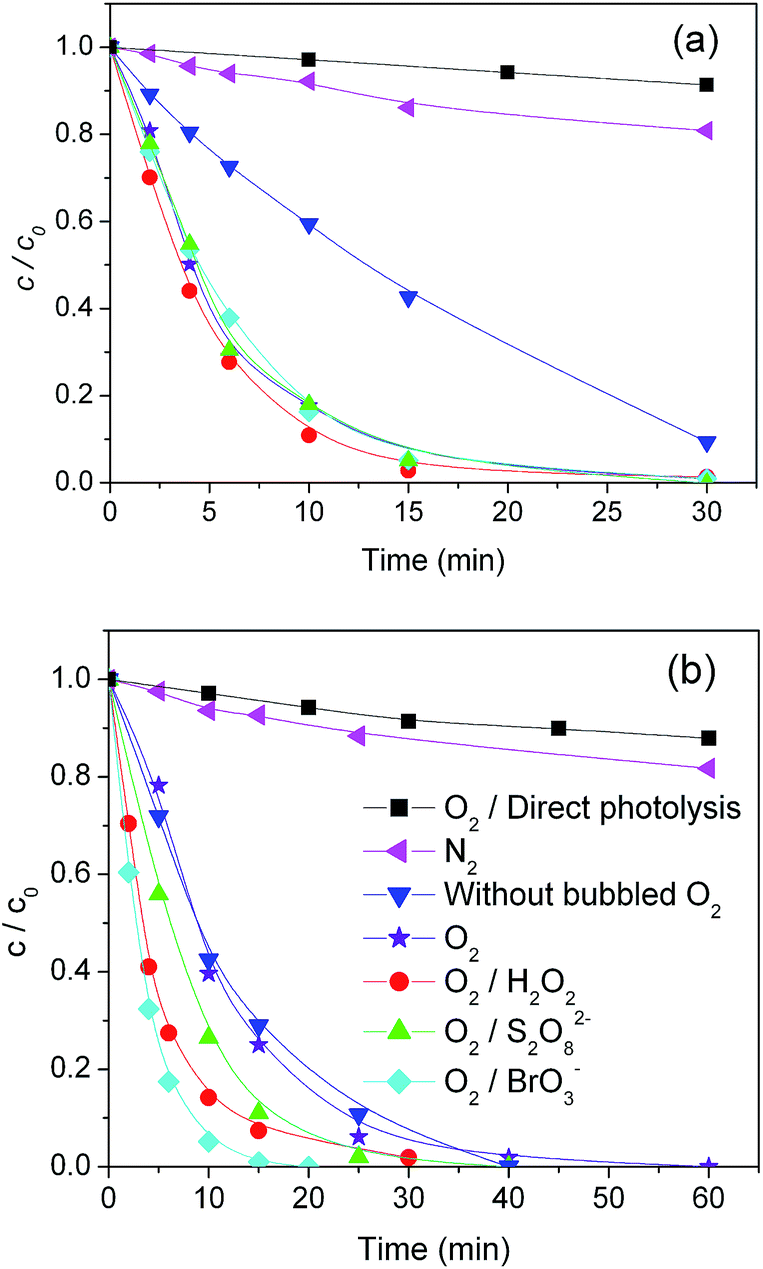 | ||
| Fig. 1 Kinetics of the photolytic/photocatalytic removal of MET using TiO2 Wackherr (a) and Degussa P25 (b) under UV irradiation, in the absence/presence of different electron acceptors. | ||
As expected, all the studied electron acceptors increased the efficiency of MET photocatalytic removal. It was determined that the influence of electron acceptors on the efficiency of degradation followed the order: O2/H2O2 > O2 ≈ O2/S2O82− ≈ O2/BrO3− using TiO2 Wackherr, and O2/BrO3− > O2/H2O2 > O2/S2O82− > O2 using Degussa P25 (Fig. 1 and Table 1). The added oxidants (H2O2, BrO3−, S2O82−) did not cause MET transformation in the dark.
Moreover, MET degradation using TiO2 under N2 atmosphere was very slow, and only marginally faster compared to the direct photolysis (MET irradiation in air without TiO2). As reported in Fig. 1 and Table 1, a considerable enhancement of MET degradation was observed, with irradiated TiO2, in air compared to N2 atmosphere: the increase of the pseudo-first order rate constant k′ was around 7 times with TiO2 Wackherr and around 12 times with Degussa P25.
Moreover, while MET degradation with Degussa P25 was little influenced by a further increase of the oxygen concentration (compare the runs with and without bubbled O2), a further significant increase of the MET rate constant (over three times) was observed with TiO2 Wackherr. Interestingly, in the case of TiO2 Wackherr the degradation of MET did not undergo a further important enhancement upon addition of other e− scavengers. In contrast, with Degussa P25 the additional effect of the scavengers was substantial. The differences between the two photocatalysts may be connected with the different surface area (much larger for Degussa P25) and different crystal structure (presence of the rutile phase in Degussa P25). These issues could lead to differences in the interaction of O2 and other e− scavengers with the photocatalyst surface. In the case of TiO2 Wackherr, it appears that O2 bubbling already brought the reaction to its optimum and that any further enhancement was difficult. The situation was very different for Degussa P25.
As far as the role of O2 as electron acceptor is concerned, photoelectrons can be captured by O2 to produce O2˙−, H2O2 (reactions (4), (7) and (8)) and then eventually hydroxyl radicals (reactions (9)–(11)):8,10,38
| O2˙− + H+ → HO2˙ | (7) |
| 2HO2˙ → H2O2 + O2 | (8) |
| H2O2 + hν → 2˙OH | (9) |
| H2O2 + e− → ˙OH + OH− | (10) |
| H2O2 + O2˙− → ˙OH + OH− + O2 | (11) |
When comparing the systems O2 and O2/H2O2 (oxygen bubbling in both cases, reported as purple stars and red circles, respectively, in Fig. 1), one can notice a small acceleration (by a factor of 1.2) caused by H2O2 addition in the case of TiO2 Wackherr, and a more marked H2O2 effect (2.2 times) with Degussa P25. Despite the more important role of H2O2, MET degradation with Degussa P25 and O2/H2O2 was still a bit slower compared to TiO2 Wackherr with otherwise identical conditions. The fact that a photocatalyst with lower surface area (TiO2 Wackherr) could induce faster MET degradation compared with Degussa P25 is most likely accounted for by lower radiation scattering, which allows a better use of the incoming photons.13
The increase of MET degradation rate upon addition of H2O2 to Degussa P25 (Table 1) was likely due to increased generation of ˙OH. Indeed, H2O2 can enhance ˙OH production through the following pathways: (I) direct photolysis of H2O2 under UV irradiation (reaction (9)); (II) reaction between H2O2 and e−: H2O2 is a more effective electron acceptor than oxygen because its reaction with e− yields ˙OH and OH− (reaction (10)), while the corresponding process with O2 produces the weaker oxidant O2˙− (reaction (4)); (III) reaction with O2˙−, also yielding ˙OH (reaction (11)).8,10,38
In the presence of Degussa P25, the O2/BrO3− system was the most efficient for the degradation of MET. In contrast, the addition of BrO3− did not have an important effect on MET degradation with TiO2 Wackherr. When operational, the enhancement of photodegradation efficiency would likely be a consequence of the reaction between BrO3− and conduction-band electrons. A first effect is the inhibition of e−–h+ recombination, which prolongs the life time of the photogenerated holes. One should additionally consider the formation of the reactive species BrO2˙ (reaction (12)).38,39 The possible role of BrO2˙ in MET degradation was investigated with DFT methods (vide infra).
| BrO3− + 2H+ + 2e− → BrO2˙ + H2O | (12) |
The addition of persulfate to the O2-bubbling system (to obtain O2/S2O82−) had a limited enhancement effect in the case of Degussa P25 (1.4 times) and practically no effect with TiO2 Wackherr. The reaction (13) between S2O82− and e− yields the strong oxidant SO4˙−, but SO42− is also formed and it can act as a hole scavenger (reaction (14)).10,38 The trade-off between h+ and SO4˙− could result into a limited enhancement effect, or even into no effect.10
| S2O82− + e− → SO42− + SO4˙− | (13) |
| SO42− + h+ → SO4˙− | (14) |
Another issue is that sulfate, formed in reaction (13), can be adsorbed on the TiO2 surface and decrease the photocatalytic activity of the oxide.10 Since S2O82− turned out to be quite ineffective as electron acceptor in the removal of MET, it was not subjected to further investigation. The very low interaction between SO4˙− and MET, compared to other systems, was confirmed through theoretical analysis as well (vide infra).
The results reported, and in particular the much faster transformation of MET with O2 compared to N2 bubbling, highlight the importance of electron acceptors in the degradation process. The addition of further electron acceptors may enhance degradation, but the reaction pathways are probably modified as well. Fig. 2 reports chromatograms obtained using TiO2 Wackherr and Degussa P25 in the presence of the studied electron acceptors, showing that the occurrence of certain intermediates (represented by unassigned peaks) significantly depended on the applied electron acceptor and catalyst. Moreover, according to literature data,10,38,40,41 it is suggested that the application of an optimal concentration of electron acceptors is of great importance to achieve the maximum removal of organic compounds from the system. For this reason, the effects of the acceptor concentrations were studied in this case as well.
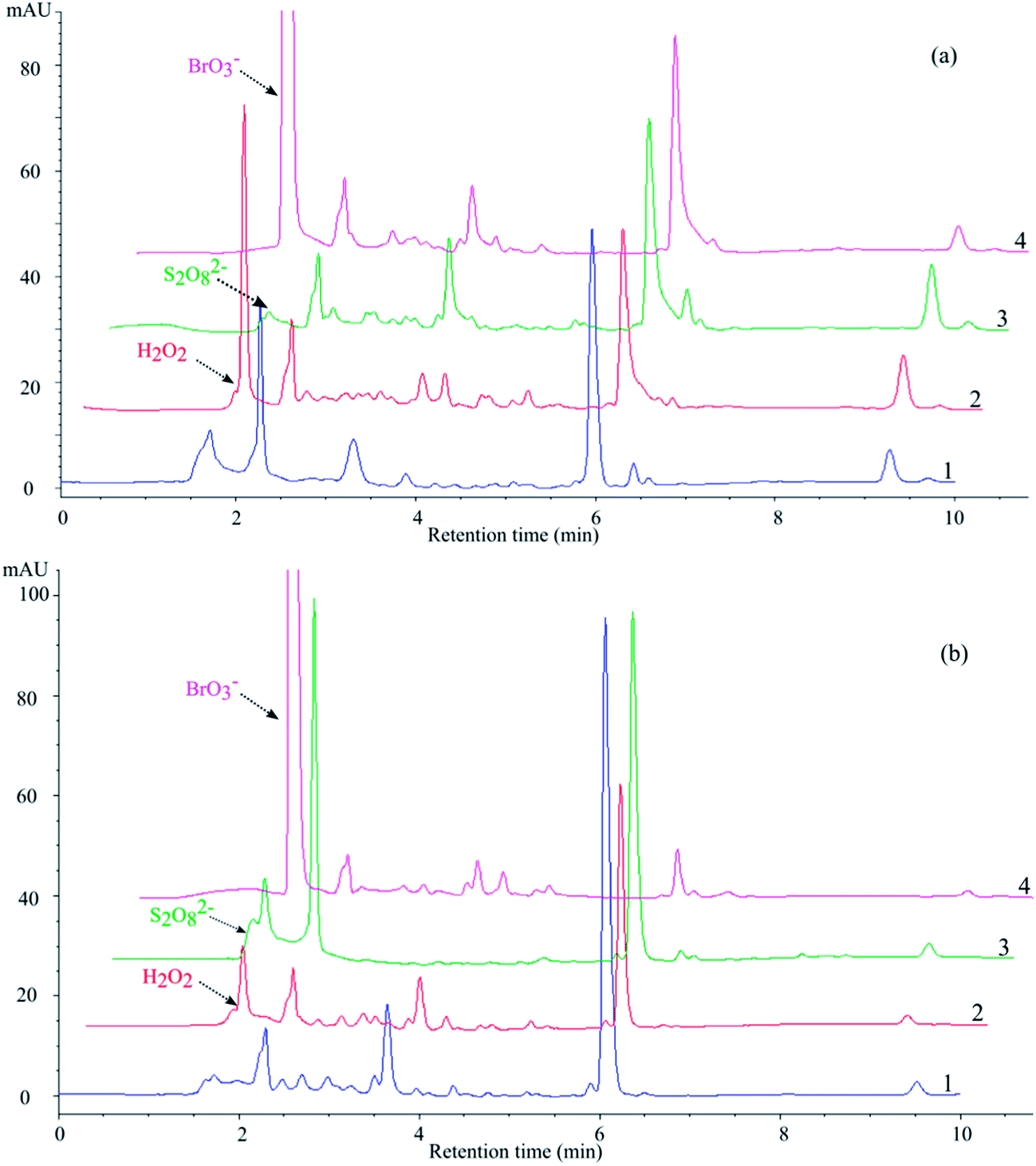 | ||
| Fig. 2 Chromatograms obtained after 10 min of MET exposure to UV irradiation in the presence of TiO2 Wackherr (a) and Degussa P25 (b), using different electron acceptors (1: O2; 2: H2O2; 3: S2O82−; 4: BrO3−). λdet = 225 nm. | ||
3.2 Effect of the initial concentration of hydrogen peroxide
The influence of the concentration of H2O2 on the efficiency of MET removal was investigated in the range of 1.0 to 5.0 mM (Fig. 3). For both Degussa P25 and TiO2 Wackherr, the optimal concentration of H2O2 was 3 mM. Under such conditions, after 10 min irradiation of the O2/H2O2 system, the degradation of MET reached 89% with TiO2 Wackherr and 86% with Degussa P25 (Fig. 3).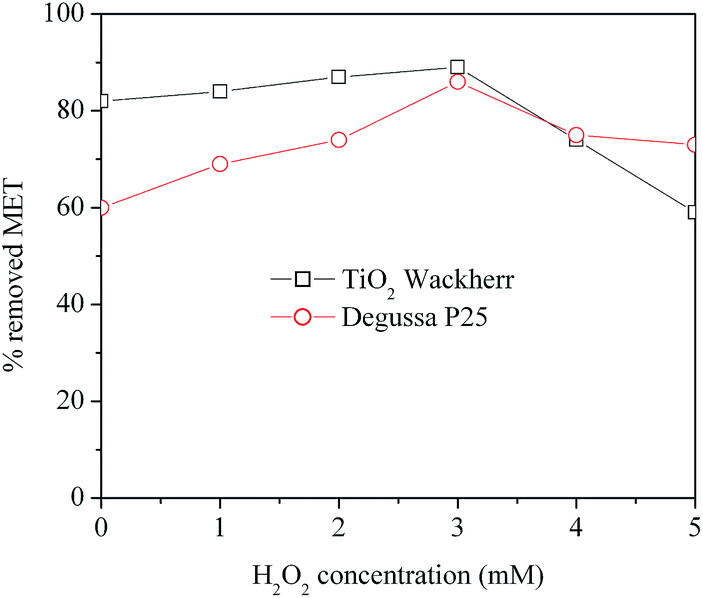 | ||
| Fig. 3 Effect of the initial concentration of H2O2 on MET removal using O2/UV/TiO2 for the first 10 min irradiation. | ||
The presence of H2O2 at concentration values above 3 mM resulted in lower MET degradation compared to the optimal conditions. The most likely reason is that H2O2 acts as an ˙OH scavenger, generating the much less reactive hydroperoxyl/superoxide radicals (HO2˙/O2˙−, reaction (15)). Moreover, HO2˙ can further react with ˙OH to form oxygen and water, which are not directly involved into MET degradation (reaction (16)).10,40
| H2O2 + ˙OH → HO2˙ + H2O (or O2˙− + H+ + H2O) | (15) |
| HO2˙ + ˙OH → O2 + H2O | (16) |
Additionally, H2O2 at elevated concentration could react with TiO2 to form peroxo compounds, which are detrimental to the photocatalytic action.42
3.3 Effect of the initial concentration of potassium bromate
Fig. 4 reports the MET degradation efficiency in the presence of different initial concentrations of BrO3−. In the case of TiO2 Wackherr, as already discussed, the bromate effect was very limited. In contrast, Degussa P25 showed a degradation increase that quickly tended to a plateau. In the latter case the most effective MET degradation was observed for 3 mM bromate, but very little difference could be detected in the 3–5 mM BrO3− concentration range. While e− scavenging and a potential involvement of BrO2˙ (formed in reaction (12)) could possibly enhance degradation, at elevated BrO3− levels the system reactivity could be limited by the formation of bromide (reaction (17)). The latter could both adsorb on the photocatalyst surface and be involved in h+ and ˙OH scavenging.10,38,41,43,44| BrO3− + 6H+ + 6e− → [BrO2−, HOBr] → Br− + 3H2O | (17) |
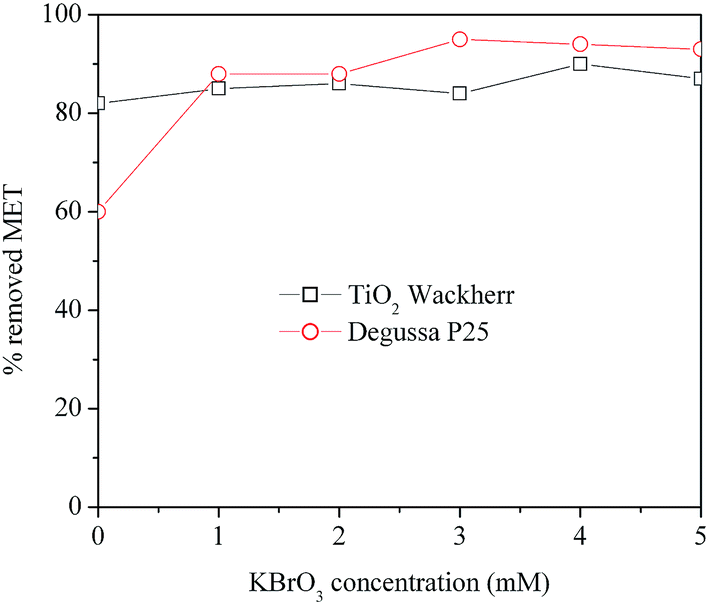 | ||
| Fig. 4 Effect of the initial concentration of BrO3− on MET removal using O2/UV/TiO2 for the first 10 min of irradiation. | ||
In the studied systems the electron acceptors are expected to inhibit the e−–h+ recombination processes, but they could have additional effects (e.g. production of reactive species by photolysis, such as ˙OH from H2O2, see reaction (9)). These effects can be highlighted in the absence of TiO2. For this reason, MET was irradiated alone and in the presence of O2/H2O2 and O2/BrO3−, without TiO2 (Fig. 5). The degradation of MET was considerably slower compared to photocatalytic conditions, but in the presence of H2O2 around 50% of MET was removed from the system after 60 minutes irradiation. In this case, acceleration of degradation compared to MET direct photolysis would probably be accounted for by ˙OH, generated by H2O2 irradiation. Indeed, the hydroxyl radical reacts rapidly and non-selectively with most organic compounds, either by H-abstraction or by addition to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C unsaturated bonds.45 Although slower compared to MET photocatalytic degradation, the use of H2O2 under irradiation could be attractive due to its simplicity.
C unsaturated bonds.45 Although slower compared to MET photocatalytic degradation, the use of H2O2 under irradiation could be attractive due to its simplicity.
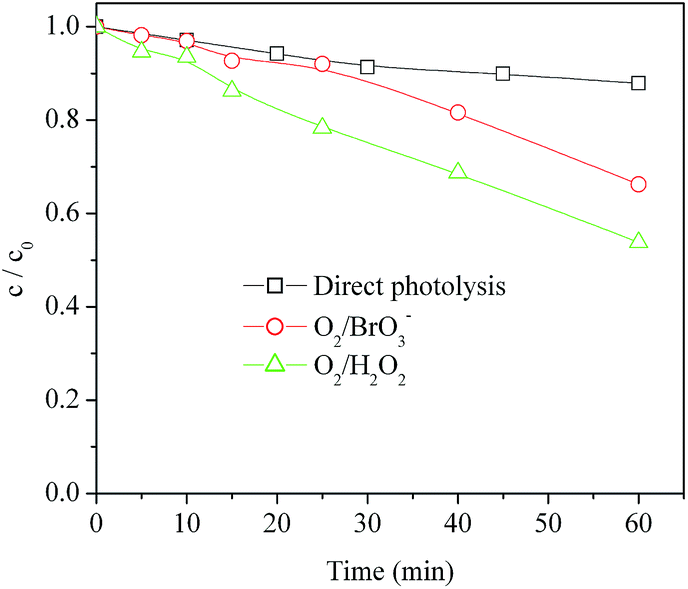 | ||
| Fig. 5 Kinetics of the direct and indirect photolysis of MET under UV irradiation in the presence of O2, O2/BrO3− and O2/H2O2. | ||
Some enhancement of MET degradation was also observed in the presence of irradiated bromate, in particular at longer irradiation times (>30 min), in agreement with literature reports.46
3.4 Evaluation of the degree of mineralization
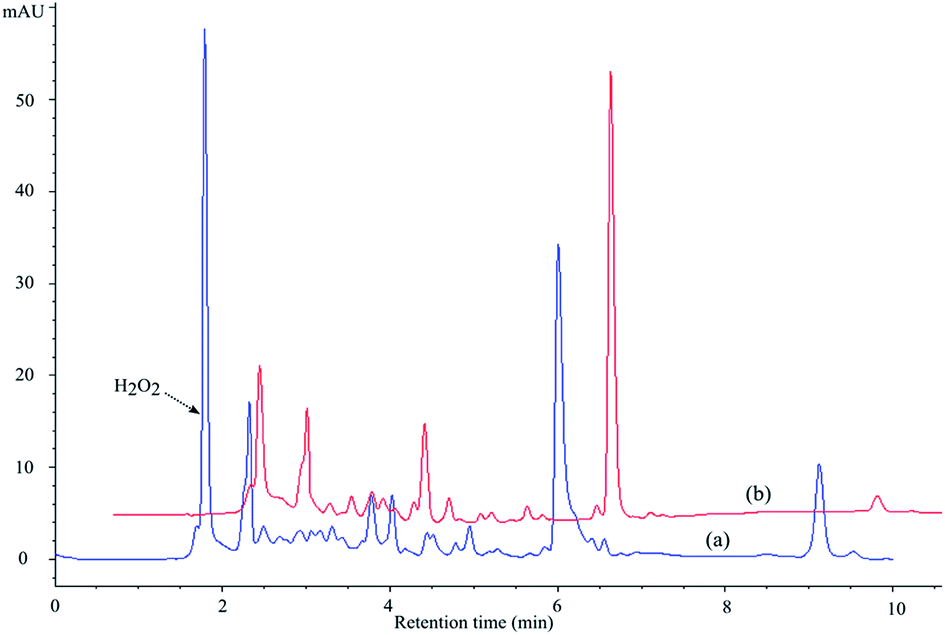 | ||
| Fig. 6 Chromatograms obtained after 10 min of MET irradiation in the presence of TiO2 Wackherr (a) and Degussa P25 (b): O2/H2O2 system in both cases (O2 bubbling, 3 mM H2O2), λdet = 225 nm. | ||
Therefore, the ability of the systems to achieve MET mineralization is particularly important in the case of the Degussa P25 photocatalyst. Further experimental results obtained in the present work indicate that the adsorption of MET on the catalysts doesn't differ more than 10%, which leads to conclusion that the adsorption of the investigated compound doesn't have significant influence on the efficiency of photocatalytic degradation. As far as mineralization is concerned, soon after complete removal of MET from the system, 88% of organic compounds (measured as organic carbon) were still present with TiO2 Wackherr in the absence of H2O2, and even 94% (that is, only 6% mineralization) with 3 mM H2O2 (Fig. 7a). The degree of mineralization after MET disappearance was significantly higher using Degussa P25, which gave 33% residual organic carbon without H2O2 and 72% with H2O2 (Fig. 7b). Photonic efficiencies48 calculated from data obtained for 60 min of mineralization of MET in the presence of TiO2 Wackherr and Degussa P25 without H2O2 were 0.068%, and 0.223%, respectively. Also, for the systems TiO2 Wackherr and Degussa P25 in the presence of H2O2, the photonic efficiency was 0.012% and 0.066%, respectively. The better performance of Degussa P25 toward mineralization, compared to TiO2 Wackherr, could be accounted for by its higher surface area. Indeed, a photocatalyst with low surface area, such as TiO2 Wackherr, could undergo surface poisoning by the degradation intermediates.13 As an alternative or in addition, the mineralization of MET could be connected with reactions involving h+. The latter are favoured in the presence of Degussa P25 compared with TiO2 Wackherr, which induces ˙OH reactions to a higher extent.13 This issue would be consistent with the previously discussed finding (Fig. 2) that different chromatographic peaks, corresponding to different intermediates, could be detected with the two photocatalysts.
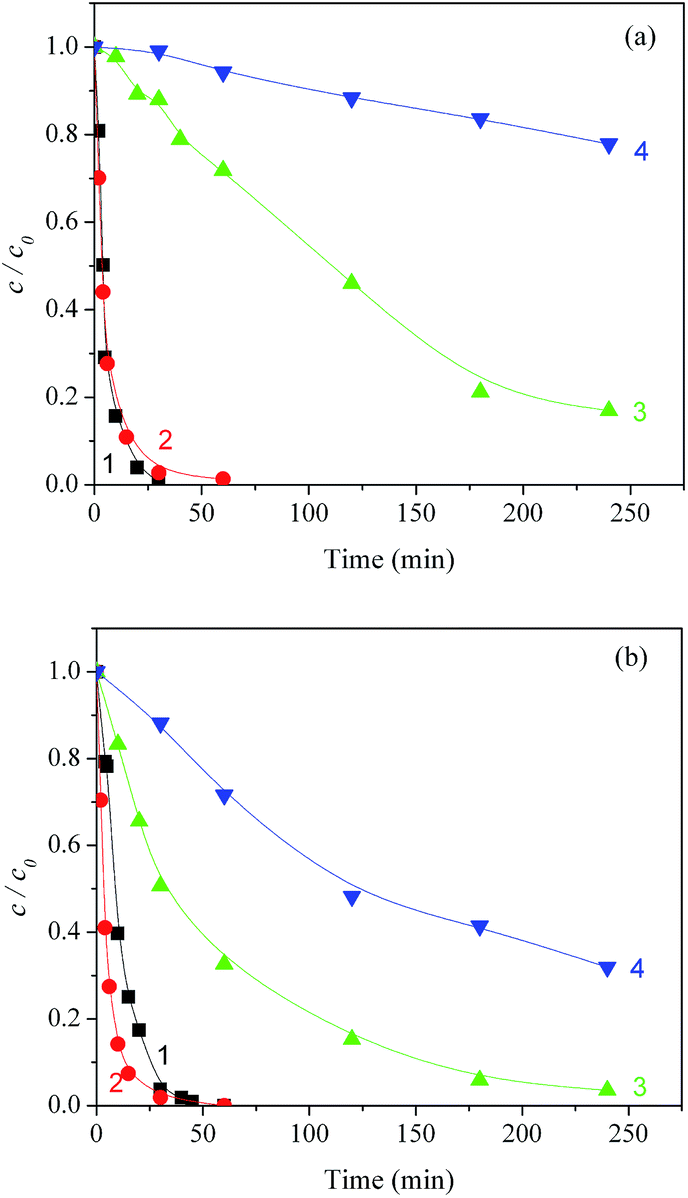 | ||
| Fig. 7 Kinetics of photocatalytic degradation of MET under UV irradiation and O2 bubbling, in the presence of TiO2 Wackherr (a) and Degussa P25 (b): (1) MET trend, no H2O2; (2) MET trend, 3 mM H2O2; (3) TOC trend, no H2O2; (4) TOC trend, 3 mM H2O2. | ||
With both TiO2 types, mineralization further increased up to the longest irradiation time (4 h). However, while H2O2 enhanced MET degradation (at least with Degussa P25), it slowed down mineralization with both photocatalysts. If the above hypothesis concerning h+vs. ˙OH is correct, the addition of H2O2 would shift the system reactivity towards the hydroxyl radical and the inhibition of mineralization would be automatically explained.
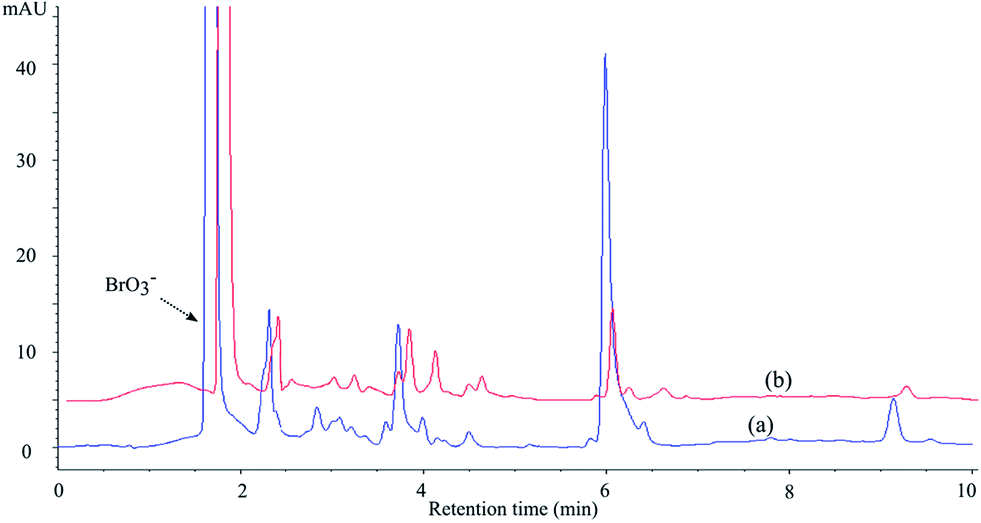 | ||
| Fig. 8 Chromatograms obtained after 10 min of UV irradiation of MET in the presence of TiO2 Wackherr (a) and Degussa P25 (b), in the O2/BrO3− system (3 mM bromate, O2 bubbling); λdet = 225 nm. | ||
TOC measurements (Fig. 9) show that bromate slightly increased mineralization with TiO2 Wackherr and slightly decreased it with Degussa P25. After 240 minutes of irradiation without BrO3−, the percentage of the residual organic compounds was reduced to 17% for TiO2 Wackherr and to 4% for Degussa P25. In the presence of BrO3−, the corresponding values were 5% for TiO2 Wackherr and 13% for Degussa P25. Photonic efficiencies of mineralization of MET after 60 min in the presence of TiO2 Wackherr and Degussa P25 with KBrO3 were 0.096%, and 0.194%, respectively.
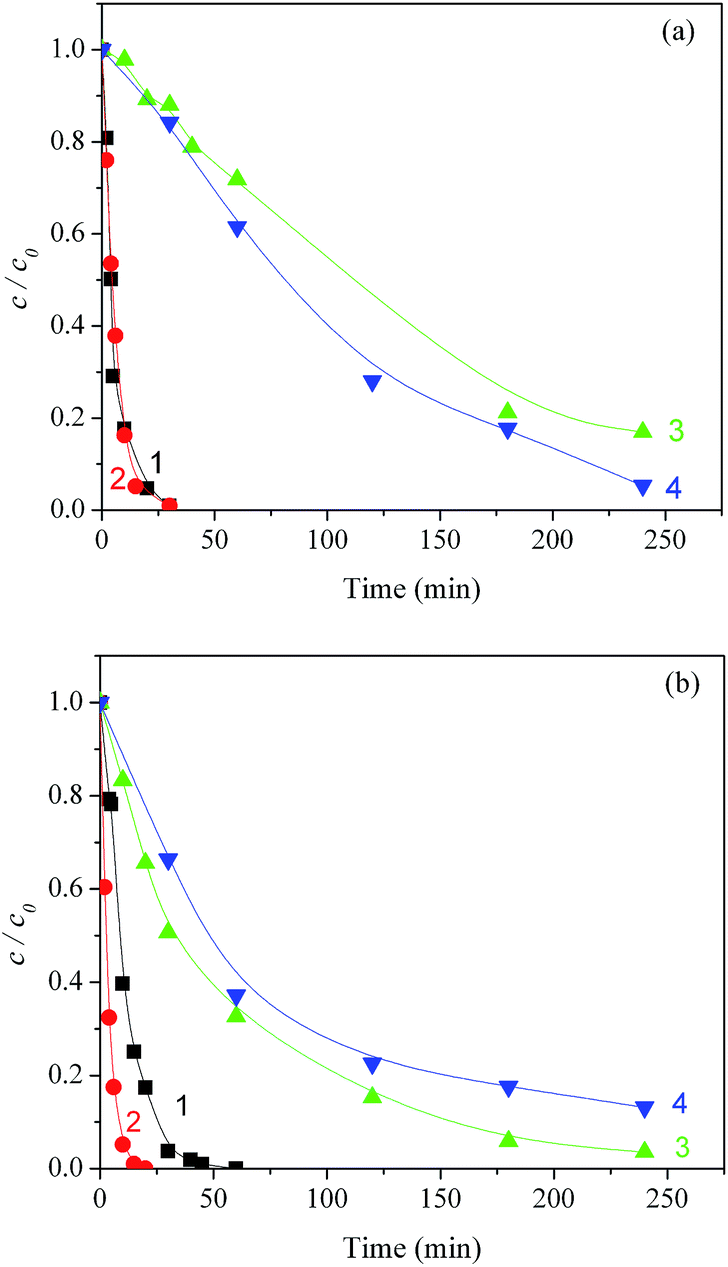 | ||
| Fig. 9 Kinetics of photocatalytic degradation of MET under UV irradiation with O2 bubbling, in the presence of TiO2 Wackherr (a) and Degussa P25 (b): (1) MET trend, no BrO3−; (2) MET trend, 3 mM BrO3−; (3) TOC trend, no BrO3−; (4) TOC trend, 3 mM BrO3−. | ||
3.5 Influence of radicals on MET-DFT insight
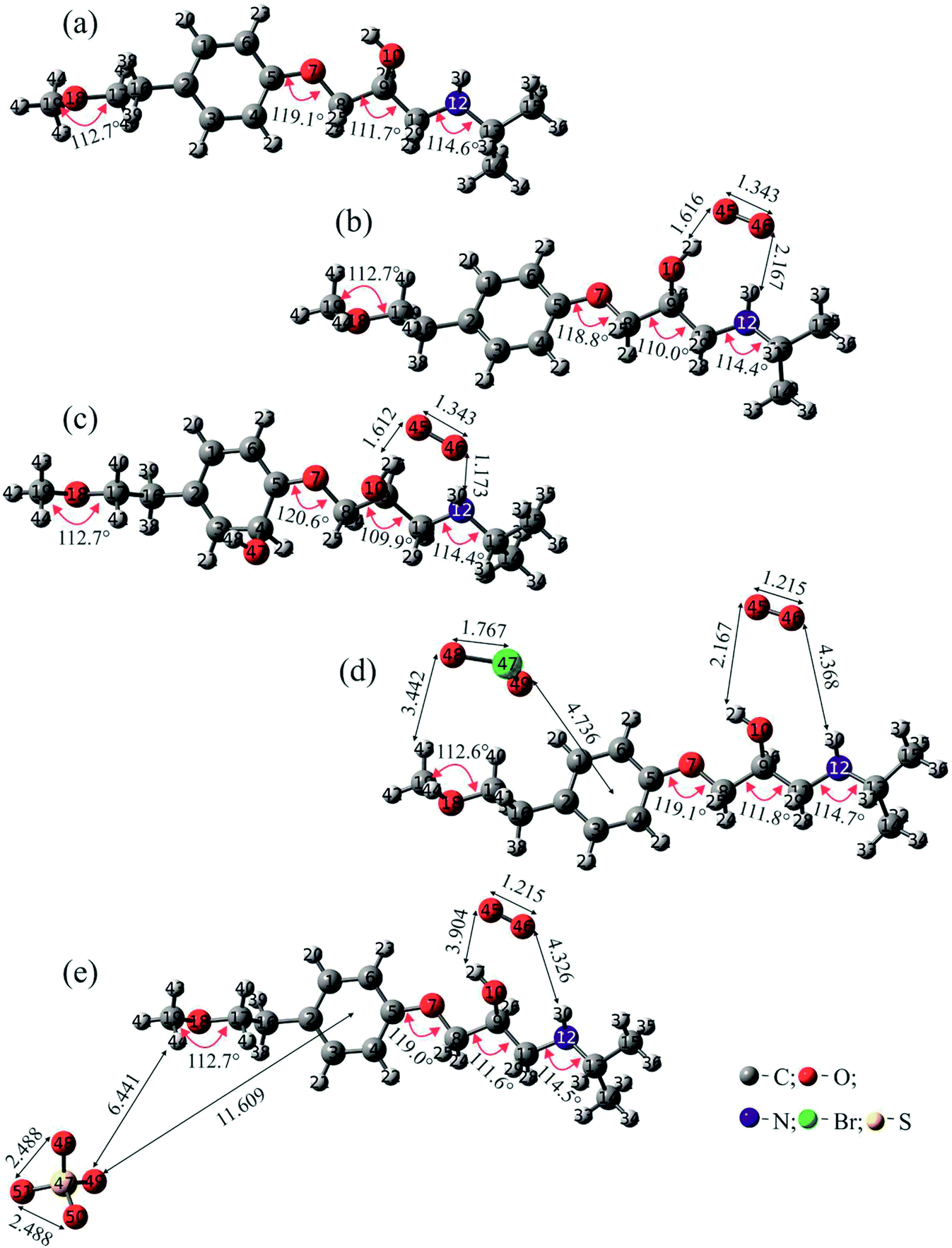 | ||
| Fig. 10 Optimized geometries of the investigated structures with specific angles (degrees) and distances (Å) for: (a) MET; (b) MET/O2˙−; (c) MET/O2˙−/˙OH; (d) MET/O2˙−/BrO2˙; and (e) MET/O2˙−/SO4˙−. | ||
The interaction distance between O2˙− and MET was the shortest (1.612 Å) when ˙OH was also present, while it was the longest (3.904 Å) in the presence of SO4˙−. Concerning other radicals, the interaction distance with MET was ca. 3.4 and 6.4 Å for BrO2˙ and SO4˙−, respectively. The most interesting situation for the interaction of radicals with MET was found in the case of ˙OH, where a new bond was formed. In various experimental studies carried out with molecules similar to MET, it was concluded that ˙OH binds to the aromatic ring.13,49 However, it was still unclear which carbon atom formed a bond with ˙OH. According to our study, a bond is formed with the carbon atom number 4 (Fig. 10).
Structural properties indicate that the highest interaction between MET and radicals takes place in the MET/O2˙−/˙OH system. These data are in overall agreement with the experimental results reported before, which emphasized the important role of O2vs. deoxygenated systems and the effect of electron acceptors in the enhancement of ˙OH (and h+) occurrence, through inhibition of e−–h+ recombination. This fact is also confirmed by the NBO analysis provided in Section 3.5.3 (vide infra).
In order to understand the reactive properties of MET, we will refer to Fukui functions and Fukui indices. Fukui functions are presented in Fig. 11, while Fukui indices (fNN HOMO and fNN LUMO) are given in Table 2. Fukui indices are commonly used since they describe the electron density when the molecule is subjected to a reaction that modifies the electron density itself.50–52 Concerning the Fukui functions (f− and f+), the red colour corresponds to the negative values while the blue colour corresponds to the positive ones. Negative values of f− correspond to regions that lose electron density when the molecule is subjected to electrophilic attack, or when the molecule itself acts as a nucleophile. Positive values of f+ correspond to areas that gain electron density when the molecule is subjected to nucleophilic attack, or when it acts as electrophile.
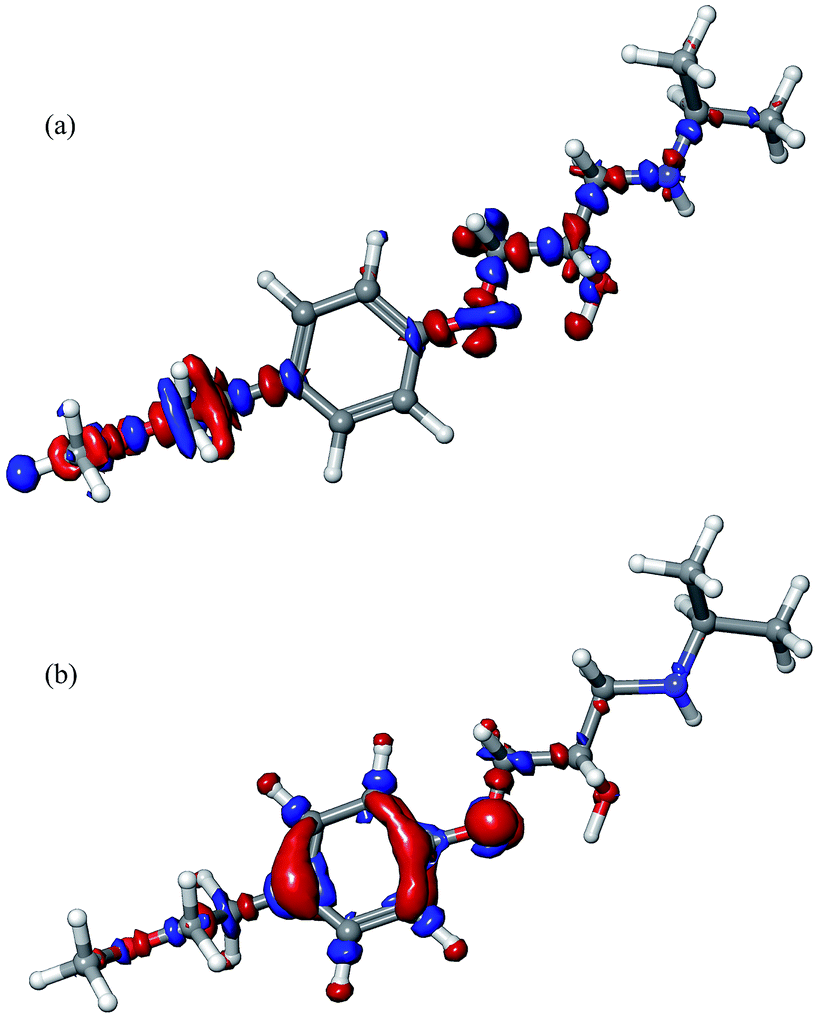 | ||
| Fig. 11 Fukui functions of MET: (a) Fukui f+ function and (b) Fukui f− function. | ||
| Atom | f NN HOMO | f NN LUMO |
|---|---|---|
| C1 | 0.0344 | 0.2021 |
| C2 | 0.2415 | 0.0115 |
| C3 | 0.0714 | 0.2885 |
| C4 | 0.1006 | 0.1777 |
| C5 | 0.1795 | 0.0070 |
| C6 | 0.1184 | 0.2969 |
| O7 | 0.1739 | 0.0006 |
| C8 | 0.0012 | 0.0014 |
| C9 | 0.0000 | 0.0048 |
| O10 | 0.0007 | −0.0026 |
| C11 | 0.0013 | 0.0011 |
| N12 | 0.0061 | −0.0008 |
| C13 | 0.0004 | 0.0002 |
| C14 | 0.0002 | 0.0001 |
| C15 | 0.0000 | 0.0001 |
| C16 | 0.0071 | 0.0016 |
| C17 | 0.0219 | −0.0035 |
| O18 | 0.0025 | 0.0004 |
| C19 | 0.0018 | 0.0004 |
The fNN HOMO indices are related to the f− Fukui function, while the fNN LUMO ones are related to f+. In other words, a high positive value of fNN HOMO indicates that the relevant atom can donate electrons, thereby acting as a nucleophile, while an elevated fNN LUMO indicates that the atom can receive electrons, thus acting as an electrophile. Having in mind the fact that a new bond was formed between MET and ˙OH, special attention was paid to the C4 atom of the MET aromatic ring, namely the atom involved in bond formation. According to positive surface of Fukui f+ function, the MET molecule has an electrophilic nature on both tail parts. On the other hand, the negative value of f− located at the aromatic ring, and automatically at the C4 atom, suggests that this atom could act as a nucleophile as well. Significant values of both Fukui functions are located at the oxygen atom O7 (Fig. 11), while its Fukui indices emphasize nucleophilic nature (Table 2).
Concerning Fukui indices, the value of fNN HOMO for the atom C4 again emphasizes its nucleophilic nature, while the highest value of this index is recorded for the C2 atom. The highest value of fNN LUMO is obtained for the atom C6. Overall, Fukui indices have significantly high values for atoms belonging to the aromatic ring.
One should be careful when interpreting the results of Fukui functions and indices, since they are not reliable to absolutely identify the most reactive electrophilic or nucleophilic sites within a particular molecule. In such a case, they can serve only as qualitative indicators of reactivity. However, according to both Fukui functions and indices, the C4 atom of the MET aromatic ring is a potential reaction site, which is confirmed in the case of the MET/O2˙−/˙OH system.
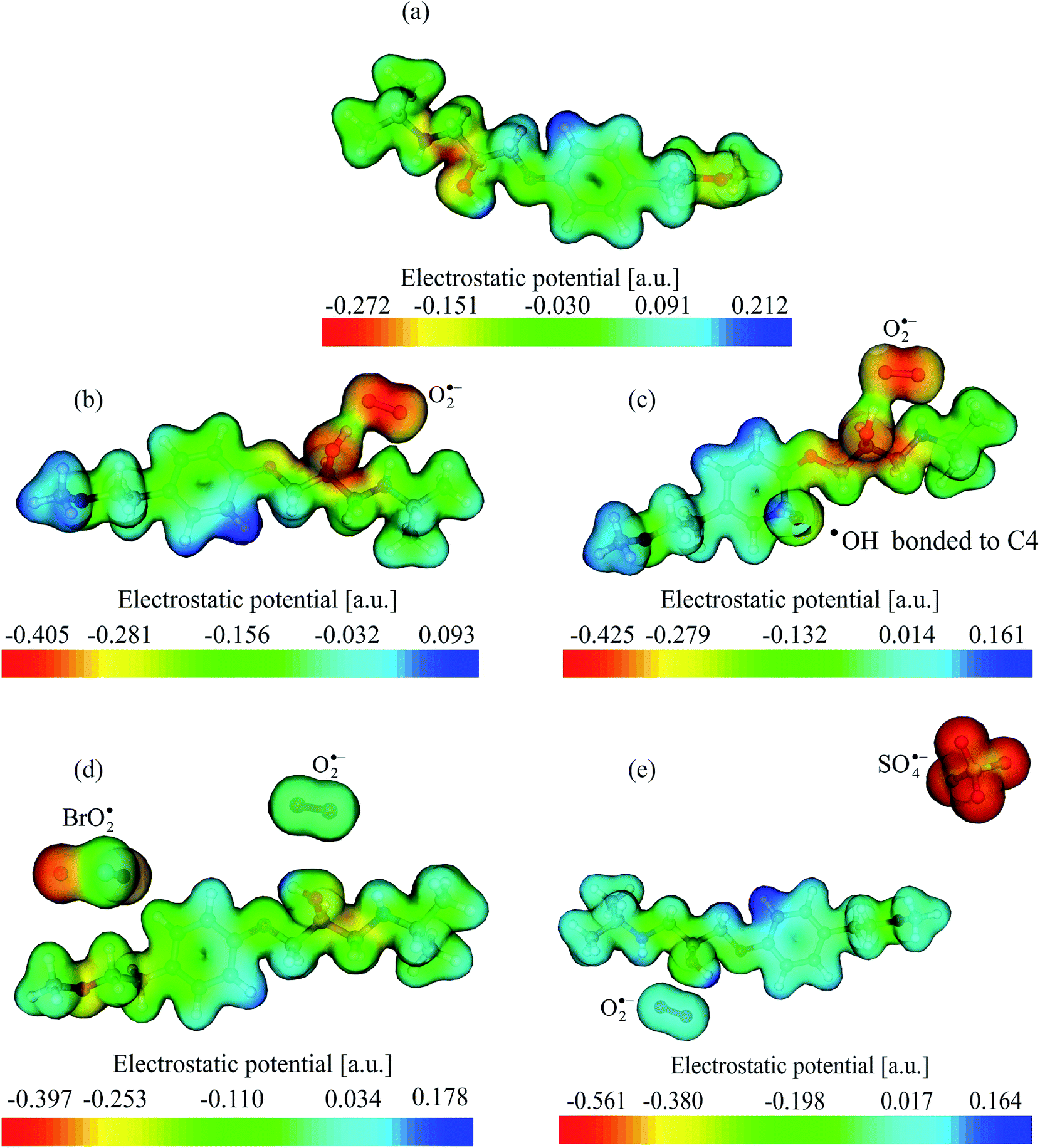 | ||
| Fig. 12 Representative MEP surfaces of the investigated systems: (a) MET; (b) MET/O2˙−; (c) MET/O2˙−/˙OH; (d) MET/O2˙−/BrO2˙; and (e) MET/O2˙−/SO4˙−. | ||
| System | Donor unit (NBOs of) | Acceptor unit (NBOs of) | ∑E(2) [kcal mol−1] |
|---|---|---|---|
| MET/O2˙− | MET | O2˙− | 3.14 |
| O2˙− | MET | 34.31 | |
| MET/O2˙−/˙OH | MET | O2˙− | 0.23 |
| MET | ˙OH | Bonded | |
| ˙OH | MET | Bonded | |
| O2˙− | MET | 28.21 | |
| O2˙− | ˙OH | <0.05 | |
| ˙OH | O2˙− | <0.05 | |
| MET/O2˙−/BrO2˙ | MET | O2˙− | <0.05 |
| MET | BrO2˙ | <0.05 | |
| O2˙− | MET | 0.64 | |
| BrO2˙ | MET | 6.08 | |
| O2˙− | BrO2˙ | <0.05 | |
| BrO2˙ | O2˙− | <0.05 | |
| MET/O2˙−/SO4˙− | MET | O2˙− | <0.05 |
| MET | SO4˙− | <0.05 | |
| O2˙− | MET | <0.05 | |
| SO4˙− | MET | <0.05 | |
| O2˙− | SO4˙− | <0.05 | |
| SO4˙− | O2˙− | <0.05 |
According to NBO results, the highest and the most important chemical interactions took place in the MET/O2˙−/˙OH system. This is consistent with above findings, which suggested that a new bond was formed in this system between the C4 atom of MET and ˙OH. The interactions resulting from electron delocalization from O2˙− to MET NBOs are significant, since ∑E(2) ≈ 28 kcal mol−1. In contrast, chemical interactions resulting from electron delocalization from MET to O2˙− NBOs are insignificant. There is a significant electron delocalization from O2˙− to MET NBOs in the case of the MET/O2˙− system, as suggested by the quite elevated ∑E(2) ≈ 34 kcal mol−1. This is the highest value of ∑E(2) among all the cases investigated in this work, and it clearly indicates a chemical interaction between O2˙− and MET. This means that dissolved oxygen, in addition to inhibiting e−–h+ recombination and enhancing, as a consequence, the occurrence and subsequent reactivity of ˙OH and h+, could further contribute to MET degradation through the formation of O2˙−.
In the case of MET/O2˙−/BrO2˙, the ∑E(2) of electron delocalization from BrO2˙ to MET NBOs is ca. 6 kcal mol−1, which suggests weak chemical interaction when compared with previous cases. The chemical interaction between O2˙− and MET in this system turned out to be insignificant, as indicated by a ∑E(2) value from O2˙− to MET of only 0.64 kcal mol−1.
While important chemical reaction between MET and BrO2˙ seems to be excluded, it should be reminded that bromate was able to significantly enhance MET photocatalytic degradation in the case of Degussa P25. In this case, the effect of bromate should probably be ascribed to its mere ability to scavenge e−, which inhibits the e−–h+ recombination and enhances the occurrence and subsequent reactivity of ˙OH and h+. Moreover, the enhancement effect with Degussa P25 combined with no bromate effect on TiO2 Wackherr suggests that an important role in MET degradation could be played by different properties of the photocatalyst, lifetime of h+ and concentration of radical species.
In the case of MET/O2˙−/SO4˙−, the interactions between MET and the corresponding radicals would just be of an electrostatic nature, as suggested by the very low values of the ∑E(2) energies, all below the default threshold of 0.05 kcal mol−1. This result excludes chemical reactivity between MET and SO4˙−, and it helps explaining the very limited effect of S2O82− on the photocatalytic degradation of MET. Indeed, electron scavenging by persulfate would be offset by drawbacks connected with SO42− formation (adsorption on the photocatalyst surface, which reduces the photocatalytic activity), while SO4˙− would not be able to favour degradation by significantly reacting with MET.
3.6 LC-ESI-MS/MS identification of degradation intermediates
By using the LC-ESI-MS/MS technique, intermediates of MET degradation in the presence of different electron acceptors (O2, O2/H2O2 and O2/KBrO3) with both catalysts were investigated (see ESI, Fig. S1–S22 and Table S1†). Intermediates formed in the absence of electron acceptors (systems with N2) were not investigated because the rate of MET degradation was very low. Therefore, the concentration of intermediates was low as well and they couldn't be identified. Because the collision-induced dissociation patterns of MET degradation were defined previously,59 it was possible to identify the detected peaks by using the product ion MS2 spectra. The retention times of all identified degradation intermediates P1–P10 (1.18–3.98 min) were shorter than that of MET (4.43 min), due to the cleavage of the molecule and the formation of polar moieties.During the degradation of MET with TiO2 Wackherr/O2, a total of seven peaks (labeled P1–P7) corresponding to degradation intermediates were detected (ESI, Table S1† and Fig. 13). Intermediate P1 represents a compound with Mmi = 133, indicating the presence of a nitrogen atom in its structure. Based on its fragmentation pattern (ESI, Table S1†) and literature data, it was concluded that this compound is 3-(propan-2-ylamino)propane-1,2-diol, already identified.13 Intermediates P2 and P3 both correspond to compounds with Mmi = 253, but with different MS2 spectra. Based on MS2 spectra and literature data13,60 it could be stated that P2 is hydroxy derivative 4-[2-hydroxy-3-(propan-2-ylamino) propoxy]benzaldehyde, that was previously identified as MET degradation product. Fragment 212 (C10H14NO4) present in P2, is formed by loss of water and isopropyl moiety, which was further dehydrated to the m/z = 177 (i.e., loss of water and methylamine group). Radjenović et al. also identified this compound and they stated that the characteristic fragment ion m/z 133 was detected in the spectrum of the parent compound and degradation products,60 which is also in agreement with our results. The herewith provided MS2 spectra of P2 (Table S1†) are in very good agreement with those previously reported.13,60 It was not possible to determine the positions of the hydroxyls, due to the small number of fragments formed in MS2 experiments. However, based on the theoretical results (Table 3, Fig. 10 and 12), we can assume that ˙OH might be bonded to the C4 atom of the aromatic ring. Fragment 116 (C6H14ON), common to many MET degradation products, and observed in the MS2 spectrum of P3, corresponds to N-(1-methylethyl)-2-oxopropan-1-aminium and indicates the intact O-bound moiety i.e. hydroxylation of either benzene ring or C2-chain. Ion 177 fragments by consecutive loss of water (m/z = 18) and ethene (m/z = 28), indicating the presence of an hydroxyethyl moiety. The presence of ion 159 fragments in MS2 spectra of MET and P3 indicates preserved aromatic ring of MET. Detailed fragmentation of MET is described in the literature, where m/z 159 corresponds to the loss of 109 (18 + 42 + 17 + 32) mass units: water, propene, ammonia are lost from the right side and methanol from the left side of the chain.61 Thus, P3 represents either 1-[4-(1-hydroxyethyl)phenoxy]-3-(propan-2-ylamino)propan-2-ol or its 2-hydroxyethyl isomer. This compound was previously identified62,63 as the 1-hydroxyethyl isomer.
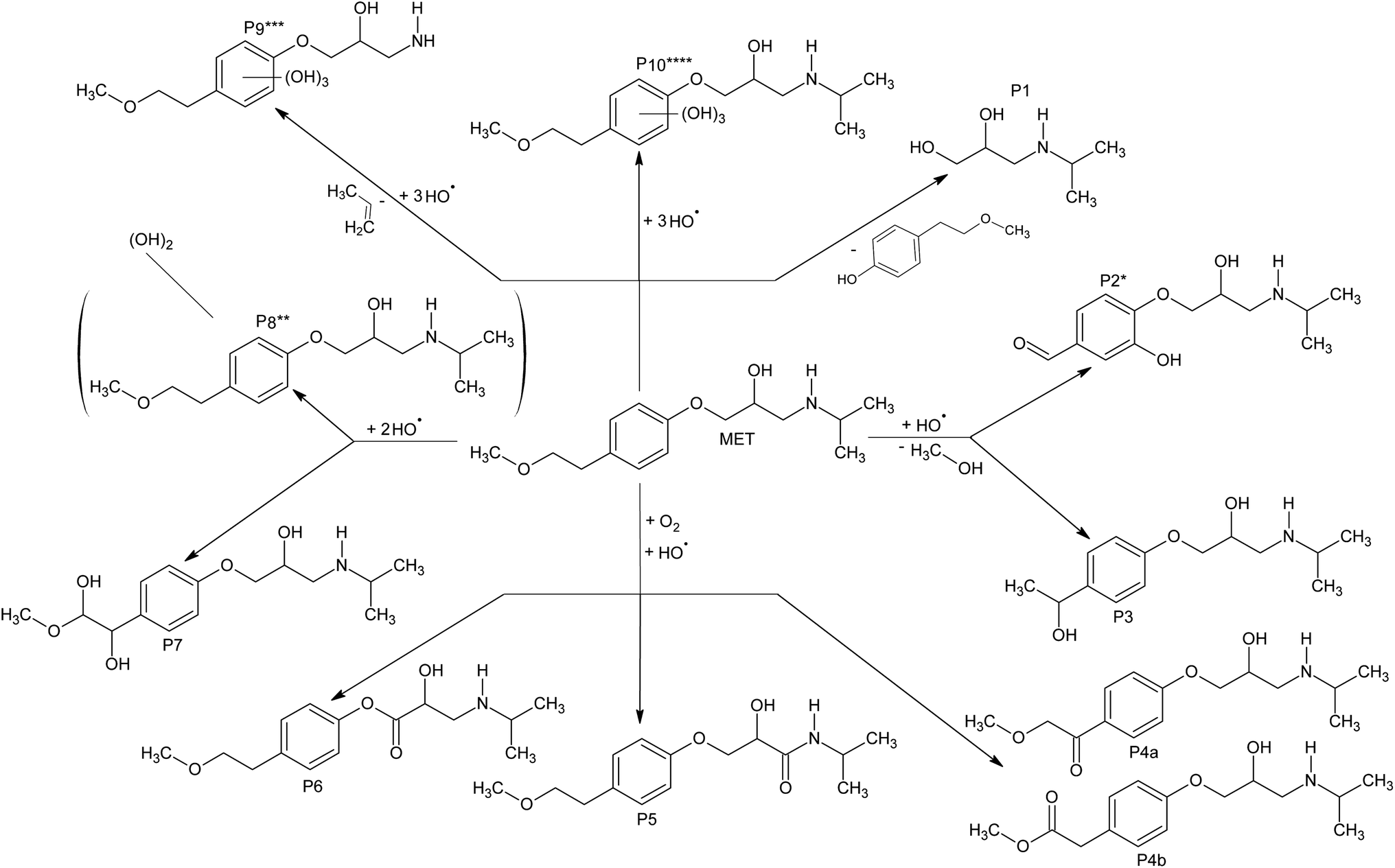 | ||
| Fig. 13 Degradation intermediates of MET with electron acceptors, in the presence of TiO2 Wackherr and Degussa P25. P1, P3–P7 were identified in all cases; P2* was identified, in all cases, only with TiO2 Wackherr; P8** was identified only with TiO2 Wackherr/O2/H2O2; P9*** was identified in all cases except TiO2 Wackherr/O2; P10**** was identified in all cases except TiO2 Wackherr/O2 and Degussa P25/O2/KBrO3. | ||
The three peaks, labelled P4, P5, and P6, corresponding to compounds with Mmi = 281, were detected. Upon further evaluation of EICs for the observed fragment ions, it was determined that peak P4 contains two closely-eluting compounds labelled P4a and P4b. Due to close elution, it was impossible to obtain pure MS2 spectra of these compounds. Therefore, EIC traces for each ion had to be checked, to confirm the presence/absence of specific fragments in each chromatographic peak. To ease the comparison of spectra and interpretation, composite MS2 spectra were prepared by summing MS2 spectra obtained at different collision energies. Molecular mass, higher by 14 units than that of MET, implies the introduction of one oxygen and the abstraction of two hydrogen atoms (i.e. either introduction of an oxo group, or introduction of hydroxyl and oxidation of the existing hydroxyl). Early loss of isopropylamine and water (Δm/z = 77, yielding fragment 205) and/or propene (Δm/z = 42, yielding fragment 240) indicate the intact iPrNH-moiety. There are five possible isomers of oxo-MET (excluding the one with oxygenated isopropyl), hereby designated A–E. Isomer A: 2-{4-[2-hydroxy-3-(propan-2-ylamino)propoxy]phenyl}ethyl formate, isomer B: methyl{4-[2-hydroxy-3-(propan-2-ylamino)propoxy]phenyl}acetate, isomer C: 1-{4-[2-hydroxy-3-(propan-2-ylamino)propoxy]phenyl}-2-methoxyethanone, isomer D: 4-(2-methoxyethyl)phenyl-2-hydroxy-3-(propan-2-ylamino)propanoate, and isomer E: 2-hydroxy-3-[4-(2-methoxyethyl)phenoxy]-N-(propan-2-yl)propanamide. Based on differences in observed spectra, we tentatively assigned structures to detected peaks. The loss of H2O (Δm/z = 18, yielding fragment 264), was observable only in peak P6, and was followed by subsequent loss of propylamine (Δm/z = 59, yielding fragment 205). In all the other peaks the latter loss is immediate, and thus only ion 205 is observable, leading to the conclusion that ion 264 is stabilized in P6. Thus, we propose that P6 represents either isomer D or E, that would – after water loss – form an α,β-unsaturated carbonyl compound, stabilized through electron delocalization. Peak P5 was characterized by the absence of two otherwise common ions for MET and derivatives: 133 (corresponding to ion produced by loss of H2O, iPrNH2 and phenyl-bound chain) and 116 (corresponding to loss of p-substituted phenoxy moiety). Fragment 177, produced by loss of 28 mass units (CO2/C2H4) from ion 205. We concluded that CO loss is to be expected if ion 205 has 1-formyl-2-[4-(2-methoxyethyl)phenoxy]ethenylium structure, i.e. if P5 could represent the isomer E (in that case, P6 would be the isomer D). The spectra of P4a and P4b differ from general pattern observed for P5 and P6 by much more pronounced fragments corresponding to loss of propene (Δm/z = 42) or water and propylamine (Δm/z = 77), and absence of otherwise common fragment 98 (either due to preferred loss of N-containing part as a neutral molecule, or due to low general abundance). Another common fragment, m/z 121, corresponding to protonated p-vinylphenol, was also absent in both peaks, which lead us to the conclusion that the oxidation possibly occurred at α- and β-position of 2-methoxyethyl moiety, i.e. that peaks P4a and P4b possibly represent isomers B and C. The presence of abundant fragment 116 (corresponding to intact N-(1-methylethyl)-2-oxopropan-1-aminium, formed by cleavage of phenoxy bond) seems to support the assumption. However, at the moment, it was not possible to determine the exact oxygenation site for these two compounds.
In the presence of H2O2 three new intermediates were observed (P8, P9 and P10), while in the presence of KBrO3 only P9 and P10 were detected. Intermediates P7 and P8 both have Mmi = 299, which is by 32 mass units greater than for MET. This implies that they are dihydroxy MET derivatives.13,49 Significant differences in fragmentation patterns of the two compounds allow the determination of the positions of the hydroxyl groups.
Intermediate P7 was identified as 1-{4-[2-hydroxy-3-(propan-2-ylamino)propoxy]phenyl}-2-methoxyethane-1,2-diol. Initial loss of 62 mass units can be attributed to cleavage of the C–C bond between CHOH and CH(OH)OCH3 units, leading to loss of methoxymethanol and formation of formyl group on benzene ring. Subsequent loss of propene (Δm/z = 42) leads to formation of ion 196. This ion further fragments by cleavage of the Ph–O bond, to yield two complementary ions at m/z 74 and 105.
Spectra of P8 are dominated by a series of fragments characteristic for MET – 159, 133, 116, 74, 72, and 56. Since these ions correspond to preserved C2–C6–O–C3–N moiety (without any additional substitution, compared to MET), we assume that the two hydroxyls are located at peripheral groups – methoxy and/or isopropyl. The value of m/z = 282 corresponds to the parent compound after loss of water. However, it was not possible to determine where one or both hydroxyls are bound, due to the lack of any other diagnostic fragments. Intermediate P9 has Mmi = 273, namely six mass units higher than MET. This is a consequence of C3H6 loss and the attachment of three hydroxyl groups. However, at lower m/z range, a series of fragments was observed – 116 (C6H14NO), 98 (C6H12N), 74 (C3H8NO), 56 (C3H6N) – corresponding to N-(1-methylethyl)-2-oxopropan-1-aminium ion and its fragment, indicating a preserved 2-hydroxy-3-[(1-methylethyl)amino]propoxy group. Further fragmentation of ion 116 into 98 by the loss of water (Δm/z = 18) supports the presumption that the three hydroxyls are bonded to the methoxyethylbenzene moiety. The absence of common ions 159, 133, and 121 supports the oxidative cleavage of benzene ring, with the loss of a C2 unit. While this reaction was previously described,63 the hereby detected compound P9 exhibit MS2 spectra different from the published ones, indicating differences in the structure and suggesting that there are three OH groups bound to a benzene ring. This leads to the conclusion that the compound is a trihydroxy derivative of 1-amino-3-[4-(2-methoxyethyl)phenoxy]propan-2-ol.
Intermediate P10, eluting at 1.57 min, corresponds to a compound with Mmi = 315. The short retention time suggests that it is very polar, which would be accounted for by the three hydroxyl groups present in its structure, since its molecular mass is 48 mass units higher than that of MET.13 The fragments 116, 98, 74, and 56 in its MS2 spectra indicate that the propanylaminopropane moiety stays intact, while the three hydroxyls are bonded to the methoxyethylbenzene part of the molecule. This compound has been previously identified as a trihydroxy derivative of 1-[4-(2-methoxyethyl)phenoxy]-3-(propan-2-ylamino)propan-2-ol.13,49
The occurrence of different intermediates under different conditions could be due to their different stabilities, and/or to differences in the degradation mechanism of MET. One issue could be the interaction of O2˙− with MET (Table 3). Compared to the case of O2 alone, the addition of H2O2 lowered such interaction by 1.2 times and that of KBrO3 by 54 times. However, NBO analysis already suggested that in the case of MET degradation with Degussa P25, besides the reactive radicals, the properties of the catalyst and the lifetime of h+ would play an important role. This issue was confirmed by the analysis of the mechanism. Namely, in the presence of Degussa P25/O2 the intermediates P1, P3–P7, P9, and P10 were identified. Moreover, they were also identified in the presence of H2O2. This issue suggests that H2O2 in this case just increased the degradation rate of the parent compound (Fig. 1), which resulted in the faster formation of the intermediates because of an increased occurrence of ˙OH. The intermediate P10 was not detected in the presence of KBrO3, which might be due to the lower ˙OH concentration in the system.
Similarly to our previous work, here it was confirmed that different sets of MET intermediates are formed with different photocatalysts.13 Indeed, P2 and P8 were not detected in the presence of Degussa P25/O2, while P8, P9 and P10 were not found with TiO2 Wackherr/O2. By comparing the O2/H2O2 systems with Degussa P25, P2 and P8 were again not identified, while in the case of TiO2 Wackherr all intermediates were identified. In the presence of O2/KBrO3 with Degussa P25 the intermediate P10 was absent, while in the presence of TiO2 Wackherr P10 was present, but P8 was absent.
In the present work, a lower number of intermediates was detected in comparison with previous work.13 That is probably a consequence of the significantly lower concentration of MET (by 60 times) used in the present work, which prevented the formation of dimeric species. Moreover, the concentrations (peak areas) of some intermediates in this work were very low and thus their identification was not possible.
4 Conclusions
This study shows that TiO2 is a very effective photocatalyst for MET degradation, in the presence of electron acceptors such as O2, H2O2, and BrO3−. The effect of the investigated electron acceptors depends on their initial concentration and on the nature of the photocatalyst. Moreover, while enhancing the transformation of MET (at least with Degussa P25), H2O2 decreased the rate of MET mineralization with both TiO2 types. This contrasting effect might be accounted for by differences in reaction pathways (h+vs. ˙OH) induced by the photocatalysts with and without H2O2, as the ˙OH pathway (enhanced by H2O2) possibly did not favour the mineralization of the substrate. The same phenomenon could also account for the higher degree of mineralization achieved, without H2O2, by Degussa P25 compared to TiO2 Wackherr. The higher surface area of Degussa P25 could also be an issue, as it would decrease the probability of photocatalyst poisoning by the degradation intermediates.DFT calculations and NBO analysis suggested that ˙OH and possibly O2˙− could undergo chemical reaction with MET. It is, therefore, suggested that dissolved oxygen would not only enhance ˙OH/h+ reactivity by scavenging e− and, therefore, inhibiting e−–h+ recombination; oxygen could additionally favour MET degradation through the formation of O2˙−. In contrast, reaction between MET and BrO2˙ or SO4˙− would be excluded. In the case of SO4˙−, the finding helps explaining the limited effect of S2O82− on MET degradation: e− scavenging by S2O82− would be largely offset by the formation of sulfate in addition to SO4˙−. While sulfate decreases the photocatalytic activity by adsorbing on the surface of TiO2, SO4˙− would not be able to take part in MET transformation.
Interestingly, both DFT calculations and the approach based on Fukui functions and Fukui indices consistently suggested that ˙OH would react with the MET aromatic ring, and particularly with its C4 atom.
Experimental study by LC-ESI-MS/MS indicated bonding of OH groups to different parts of MET, while results of theoretical analysis obtained in this investigation indicated bonding of OH group to the aromatic ring. According to the theoretical results, bonding of OH group to the C4 atom of benzene ring of P2 was suggested. It was shown experimentally that the binding of OH groups does not occur on the propanylaminopropane group chain (P8, P9, and P10) and that ring opening doesn't occur as well, which is also in agreement with theoretical analysis. Besides, intermediates that are peculiar for the systems containing H2O2 and KBrO3 were identified as well. Namely, P8, P9, and P10 were detected with TiO2 Wackherr/O2/H2O2 and P9 and P10 with TiO2 Wackherr/O2/KBrO3. While the same intermediates were identified with Degussa P25/O2 and Degussa P25/O2/H2O2, the intermediate P10 was not identified with Degussa P25/O2/KBrO3.
Acknowledgements
The authors greatly appreciate the financial support from the Ministry of Education, Science and Technological Development of the Republic of Serbia (Project no. 172042).References
- S. N. Frank and A. J. Bard, J. Phys. Chem., 1977, 81, 1484–1488 CrossRef CAS.
- P. A. Brugger, P. Cuendet and M. Graetzel, J. Am. Chem. Soc., 1981, 103, 2923–2927 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS.
- S. Ahmed, M. Rasul, W. N. Martens, R. Brown and M. Hashib, Water, Air, Soil Pollut., 2011, 215, 3–29 CrossRef CAS.
- S. Malato, P. Fernández-Ibáñez, M. Maldonado, J. Blanco and W. Gernjak, Catal. Today, 2009, 147, 1–59 CrossRef CAS.
- C. Karunakaran and R. Dhanalakshmi, Sol. Energy Mater. Sol. Cells, 2008, 92, 588–593 CrossRef CAS.
- W. Liu, S. Chen, W. Zhao and S. Zhang, Desalination, 2009, 249, 1288–1293 CrossRef CAS.
- N. Kashif and F. Ouyang, J. Environ. Sci., 2009, 21, 527–533 CrossRef CAS.
- M. Muruganandham and M. Swaminathan, Dyes Pigm., 2006, 68, 133–142 CrossRef CAS.
- N. N. Chipanina, N. F. Lazareva, T. N. Aksamentova, A. Y. Nikonov and B. A. Shainyan, Organometallics, 2014, 33, 2641–2652 CrossRef CAS.
- S. Kheirjou, A. Fattahi and M. M. Hashemi, Comput. Theor. Chem., 2014, 1036, 51–60 CrossRef CAS.
- B. Abramović, S. Kler, D. Šojić, M. Laušević, T. Radović and D. Vione, J. Hazard. Mater., 2011, 198, 123–132 CrossRef PubMed.
- S. Armaković, S. J. Armaković, J. P. Šetrajčić and I. J. Šetrajčić, J. Mol. Model., 2012, 18, 4491–4501 CrossRef PubMed.
- R. G. Parr and W. Yang, J. Am. Chem. Soc., 1984, 106, 4049–4050 CrossRef CAS.
- K. Chandrakumar and S. Pal, Int. J. Mol. Sci., 2002, 3, 324–337 CrossRef CAS.
- S. H. Rosline Sebastian Sr, M. I. Attia, M. S. Almutairi, A. A. El-Emam, C. Y. Panicker and C. Van Alsenoy, Spectrochim. Acta, Part A, 2014, 132, 295–304 CrossRef CAS PubMed.
- S. Armaković, S. J. Armaković, J. P. Šetrajčić and I. J. Šetrajčić, Chem. Phys. Lett., 2013, 578, 156–161 CrossRef.
- E. Kose, A. Atac, M. Karabacak, C. Karaca, M. Eskici and A. Karanfil, Spectrochim. Acta, Part A, 2012, 97, 435–448 CrossRef CAS PubMed.
- M. Chen, U. Waghmare, C. Friend and E. Kaxiras, J. Chem. Phys., 1998, 109, 6854–6860 CrossRef CAS.
- A. J. Rossini, R. W. Mills, G. A. Briscoe, E. L. Norton, S. J. Geier, I. Hung, S. Zheng, J. Autschbach and R. W. Schurko, J. Am. Chem. Soc., 2009, 131, 3317–3330 CrossRef CAS PubMed.
- J. Autschbach, S. Zheng and R. W. Schurko, Concepts Magn. Reson., Part A, 2010, 36, 84–126 CrossRef.
- R. J. Xavier and E. Gobinath, Spectrochim. Acta, Part A, 2012, 86, 242–251 CrossRef CAS PubMed.
- M. Snehalatha, C. Ravikumar, I. Hubert Joe, N. Sekar and V. S. Jayakumar, Spectrochim. Acta, Part A, 2009, 72, 654–662 CrossRef CAS PubMed.
- J. M. Dimitrić Marković, Z. S. Marković, J. B. Krstić, D. Milenković, B. Lučić and D. Amić, Vib. Spectrosc., 2013, 64, 1–9 CrossRef.
- Z. Marković, D. Milenković, J. Đorović, J. Dimitrić Marković, B. Lučić and D. Amić, Monatsh. Chem., 2013, 144, 803–812 CrossRef.
- S. F. Owen, E. Giltrow, D. B. Huggett, T. H. Hutchinson, J. Saye, M. J. Winter and J. P. Sumpter, Aquat. Toxicol., 2007, 82, 145–162 CrossRef CAS PubMed.
- A. M. Stolker, W. Niesing, E. Hogendoorn, J. M. Versteegh, R. Fuchs and U. T. Brinkman, Anal. Bioanal. Chem., 2004, 378, 955–963 CrossRef CAS PubMed.
- B. Kasprzyk-Hordern, R. M. Dinsdale and A. J. Guwy, Water Res., 2009, 43, 363–380 CrossRef CAS PubMed.
- D. Bendz, N. A. Paxéus, T. R. Ginn and F. J. Loge, J. Hazard. Mater., 2005, 122, 195–204 CrossRef CAS PubMed.
- S. Wiegel, A. Aulinger, R. Brockmeyer, H. Harms, J. Löffler, H. Reincke, R. Schmidt, B. Stachel, W. von Tümpling and A. Wanke, Chemosphere, 2004, 57, 107–126 CrossRef CAS PubMed.
- D. Vione, C. Minero, V. Maurino, M. E. Carlotti, T. Picatonotto and E. Pelizzetti, Appl. Catal., B, 2005, 58, 79–88 CrossRef CAS.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, J. Montgomery Jr, T. Vreven, K. Kudin and J. Burant, Gaussian 03 Search PubMed.
- A. D. Bochevarov, E. Harder, T. F. Hughes, J. R. Greenwood, D. A. Braden, D. M. Philipp, D. Rinaldo, M. D. Halls, J. Zhang and R. A. Friesner, Int. J. Quantum Chem., 2013, 113, 2110–2142 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- U. Varetto, Molekel 5.4, Swiss National Supercomputing Centre, Manno, Switzerland, 2009 Search PubMed.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- S. Chen and Y. Liu, Chemosphere, 2007, 67, 1010–1017 CrossRef CAS PubMed.
- N. A. Mir, M. M. Haque, A. Khan, M. Muneer and C. Boxall, Sci. World J., 2012, 2012, 1–8 CrossRef PubMed.
- W. Chu and C. Wong, Water Res., 2004, 38, 1037–1043 CrossRef CAS PubMed.
- V. N. Despotović, B. F. Abramović, D. V. Šojić, S. J. Kler, M. B. Dalmacija, L. J. Bjelica and D. Z. Orčić, Water, Air, Soil Pollut., 2012, 223, 3009–3020 CrossRef.
- I. Poulios, M. Kositzi and A. Kouras, J. Photochem. Photobiol., A, 1998, 115, 175–183 CrossRef CAS.
- N. San, A. Hatipoǧlu, G. Koçtürk and Z. Çınar, J. Photochem. Photobiol., A, 2001, 139, 225–232 CrossRef CAS.
- S. Qourzal, N. Barka, M. Tamimi, A. Assabbane and Y. Ait-Ichou, Appl. Catal., A, 2008, 334, 386–393 CrossRef CAS.
- M. I. Stefan, A. R. Hoy and J. R. Bolton, Environ. Sci. Technol., 1996, 30, 2382–2390 CrossRef CAS.
- N. A. Mir, A. Khan, A. Dar and M. Muneer, International Journal of Innovative Research in Science, Engineering and Technology, 2014, 3, 9333–9348 Search PubMed.
- D. D. Četojević-Simin, S. J. Armaković, D. V. Šojić and B. F. Abramović, Sci. Total Environ., 2013, 463, 968–974 CrossRef PubMed.
- D. V. Šojić, D. Z. Orčić, D. D. Četojević-Simin, V. N. Despotović and B. F. Abramović, J. Mol. Catal. A: Chem., 2014, 392, 67–75 CrossRef.
- H. Yang, T. An, G. Li, W. Song, W. J. Cooper, H. Luo and X. Guo, J. Hazard. Mater., 2010, 179, 834–839 CrossRef CAS PubMed.
- E. Chamorro and P. Pérez, J. Chem. Phys., 2005, 123, 114107 CrossRef PubMed.
- R. R. Contreras, P. Fuentealba, M. Galván and P. Pérez, Chem. Phys. Lett., 1999, 304, 405–413 CrossRef CAS.
- A. D. Bochevarov, E. Harder, T. F. Hughes, J. R. Greenwood, D. A. Braden, D. M. Philipp, D. Rinaldo, M. D. Halls, J. Zhang and R. A. Friesner, Int. J. Quantum Chem., 2013, 113, 2110–2142 CrossRef CAS.
- S. Armaković, S. J. Armaković, J. P. Šetrajčić, S. K. Jaćimovski and V. Holodkov, J. Mol. Model., 2014, 20, 1–14 Search PubMed.
- O. E. Kasende, A. Matondo, M. Muzomwe, J. T. Muya and S. Scheiner, Comput. Theor. Chem., 2014, 1034, 26–31 CrossRef CAS.
- D. Y. Buissonneaud, T. van Mourik and D. O'Hagan, Tetrahedron, 2010, 66, 2196–2202 CrossRef CAS.
- S. Armaković, S. J. Armaković and J. P. Šetrajčić, Int. J. Hydrogen Energy, 2013, 38, 12190–12198 CrossRef.
- P. R. Olivato, J. M. Santos, B. Contieri, C. R. Cerqueira, D. N. Rodrigues, E. Vinhato, J. Zukerman-Schpector and M. D. Colle, Molecules, 2013, 18, 7492–7509 CrossRef CAS PubMed.
- P. R. Olivato, J. M. M. Santos, C. R. Cerqueira Jr, E. Vinhato, J. Zukerman-Schpector, S. W. Ng, E. R. T. Tiekink and M. D. Colle, J. Mol. Struct., 2012, 1028, 97–106 CrossRef.
- R. M. Borkar, B. Raju, R. Srinivas, P. Patel and S. K. Shetty, Biomed. Chromatogr., 2012, 26, 720–736 CrossRef CAS PubMed.
- J. Radjenović, C. Sirtori, M. Petrović, D. Barceló and S. Malato, Appl. Catal., B, 2009, 89, 255–264 CrossRef.
- C. Slegersa, A. Maquillea, V. Deriddera, E. Sonveauxb, J. H. Jiwanc and B. Tilquin, Radiat. Phys. Chem., 2006, 75, 977–989 CrossRef.
- D. Šojić, V. Despotović, D. Orčić, E. Szabó, E. Arany, S. Armaković, E. Illés, K. Gajda-Schrantz, A. Dombi, T. Alapi, E. Sajben-Nagy, A. Palágyi, Cs. Vágvölgyi, L. Manczinger, L. Bjelica and B. Abramović, J. Hydrol., 2012, 472–473, 314–327 CrossRef.
- J. Benner and T. A. Ternes, Environ. Sci. Technol., 2009, 43, 5472–5480 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Influence of electron acceptors on the kinetics of metoprolol photocatalytic degradation in TiO2 suspension. A combined experimental and theoretical study. See DOI: 10.1039/c5ra10523d |
| This journal is © The Royal Society of Chemistry 2015 |