
Photophysics of crystal violet lactone in reverse micelles and its dual behaviour†
Banibrata Maity,
Aninda Chatterjee,
Sayeed Ashique Ahmed and
Debabrata Seth*
Department of Chemistry, Indian Institute of Technology Patna, Patna 800013, Bihar, India. E-mail: debabrata@iitp.ac.in; Fax: +91-612-2277383
First published on 8th June 2015
Abstract
In this article, the photophysics of Crystal Violet Lactone (CVL) in aqueous and non-aqueous reverse micelles (RMs) have been explored using UV-vis absorption, steady-state emission and picosecond time-resolved fluorescence emission spectroscopic techniques. CVL exhibits dual emission bands (CTA and CTB states of CVL) in a polar aprotic solvent, whereas when this molecule is entrapped in the RMs, it exhibits a single emission band (CTA). In the case of all the RMs, the CTA → CTB transformation is retarded due to the influence of polarity, viscosity and proticity. Moreover, in the case of viscous solvents (ethylene glycol and glycerol), a single emission band is found. The hydrogen-bond-donating solvent molecule also quenches the CTB emission band of CVL. This is the first report on the photophysics of the CVL molecule in biomimicking organised assemblies, in which the opposite spectral features were observed in the RMs at different excitation wavelengths. The average rotational relaxation time increases in the respective RMs compared to their respective solvent, with the exception of neat glycerol, wherein the rotation of CVL is practically hindered. In the case of EG and glycerol RMs, the rotational relaxation is even more retarded due to the gradual swelling of the RM cores. Herein, the hydrodynamic friction, coupled with the dielectric friction, causes such rotational motion to be quite slow.
1. Introduction
Many physicochemical and biological phenomena take place in confined media, rather than in homogeneous solution environments. Molecular confinement causes fascinating phenomena in terms of organisation and dynamics, which are completely different compared to behaviour in the bulk solution. Reverse micelles (RMs) can be considered as one of the model systems of such convenient membrane-mimetic media, which solubilize organic fluorophores, proteins, and nucleic acids without the modification of their isotropic nature.1,2 RMs have widespread applications in biological environments such as cell membranes and protein pockets.3–7 Moreover, this biomimetic system has also been used in drug delivery, enzymatic reactions, chemical catalysis, polymer synthesis and several different applications in various fields of science and technology.8–14Reverse micelles are formed by the nanoshaped self-organised aggregation of surfactants in non-polar solvents. The hydrophilic polar head groups of the surfactant molecules point inward towards the central solvent pool, while the hydrophobic, nonpolar tails protrude towards the bulk oil like nonpolar solvents. The polar solvent inside the RM cores shows different behaviour compared to bulk solvents. The size of the RM cores is characterised by w = [polar solvent]/[surfactant]. AOT (sodium 1,4-bis-2-ethylhexylsulfosuccinate) is the most commonly used surfactant in the preparation of RMs. AOT molecules have a tendency to form aggregates in the presence of non-polar solvents. The polar head groups (SO32−) of AOT surfactants are pointed towards the interior of the RMs. As a result, they are shielded from the interactions with the bulk non-polar solvent. The addition of polar solvents to an isooctane/AOT mixture forms a stable, optically isotropic solution and nanometer-sized droplets of polar solvent molecules dispersed in a nonpolar solvent, known as a microemulsion.15–17 AOT molecules have a tendency to solubilize large amounts of water molecules, depending on the size of the solvent pool, surrounding non-polar medium and temperature.18–20 The most remarkable property of RMs is their tendency to encapsulate polar solvent molecules in their oil-like nonpolar environments. However, they are practically immiscible in their bulk phase. The most common example is the water molecule. Some polar organic solvent molecules have a tendency to encapsulate in the RM core and remain dispersed in non-polar solvents. Such examples of non-aqueous polar solvents are dimethylformamide (DMF),15,16,21–23 ethylene glycol (EG),15,16,22–26 and glycerol (GY).15,16,22,23,27 Non-aqueous RMs swell to a greater extent than aqueous RMs.15,21,24,27 The polar solvent molecules inside the RM cores are highly structured.
Crystal Violet Lactone (CVL) is a photoinduced intramolecular charge transfer forming molecule, containing electron donor and acceptor moieties. This type of photoinduced electron transfer (PET) forming fluorophore has attracted increasing interest in the scientific community, due to its biological and chemical energy conversions.28,29 In the presence of proton-donating solvents, the CVL cation is formed via the ground state photodissociation of the C–O bond of the lactone ring in the CVL molecule.30,31 Owing to its labile C–O bonds, the CVL molecule has found numerous applications such as a photopolymerization initiator and an effective material in thermochromic coatings.32,33 The photophysics of organic molecules that exhibit intramolecular charge transfer (ICT) phenomena has drawn increasing attention due to their extensive application in fluorescence sensors, markers and “switches” in materials science and in biology. The formation of the ICT state can sometimes result in a dual fluorescence phenomenon, in which normal emission occurs from the locally excited state (LE), followed by additional emission from the low energy band corresponding to the ICT state. The dynamics of the charge transfer (CT) processes are mainly governed by solute–solvent interactions and excited state structural relaxation of the solute molecules. The dual fluorescence emission phenomenon is associated with intramolecular electron transfer (ET), along with excited state structural relaxation via a photoinduced adiabatic reaction mechanism.29 Spiro carbon atoms, such as those in the lactone form of rhodamine molecules (LRs) also exhibit dual fluorescence emission.34 This type of dual emission basically arises from a high charge transfer (CT) state and zwitterion formation occurs via excited state adiabatic photodissociation of the C–O bond in a phthalide ring.34 The CVL molecule also shows dual emission behaviour due to excited state charge transfer in the presence of moderately and highly polar solvent molecules.30 CVL and its derivatives emit spectrally continuous dual fluorescence-exhibiting white light and are used in organic light-emitting diodes.35 The CVL molecule consists of lactone forms of the triarylmethane moiety (LTAM), in which three individual electron-donating dialkyl aniline moieties form a propeller-like structure with the electron-accepting five membered phthalide ring. The tetrahedral sp3 carbon atom of the CVL molecule is attached to the electron donor and acceptor moieties. Due to the presence of both electron donating and electron accepting moieties, the charge transfer state of the CVL molecule is localised on the 6-dimethylaminophthalide (6-DMAPd) and the malachite green lactone (MGL) subunits. Karpiuk30 proposed that the photophysical properties of the CVL molecule undergo remarkable changes, depending on the polarity of the solvent medium. In non-polar solvents, the first excited state (charge transfer nature CTA, μ = 10.7 D) of the CVL molecule is mainly located in the DMAP subunit, which is low in energy compared to the charge transfer state (CTB, μ = 25.2 D) localised on the MGL subunit. As a result, the CVL molecule emits a single fluorescence emission band from the 6-DMAPd moiety. On the contrary, dual emission bands for the CVL molecule are observed in the presence of aprotic solvents (moderately and highly polar solvent molecules) due to reversal differences between the dipolar characters of the two emissive charge transfer states (μCTB < μCTA). Herein, the charge transfer A band (CTA) of the CVL molecule is formed from the polar excited state (localised in the 6-DMAPd subunit), whereas the charge transfer B band (CTB) is formed from the highly polar excited state via electron transfer (ET) of one of the dimethyl aniline groups to the 6-DMAPd subunit. The charge transfer CTB state populates during solvation in a very fast excited-state process.30 In the presence of protic solvents, the crystal violet cation is formed via the dissociation of the C–O bond of the lactone moiety in the CVL molecule. In the presence of alcoholic solvents, CVL exhibits a single fluorescence emission band.30 Due to hydrogen-bonding interactions between CVL and alcoholic solvent molecules, the charge transfer band (CTB) diminishes, leading to non-fluorescent behaviour by the CVL molecule in protic solvents. Samanta and co-workers36 also reported that CVL displays a single fluorescence emission band (CTA state) in the presence of some room temperature ionic liquids (RTILs). The charge transfer CTB band is completely prevented and the CTA → CTB transformation is retarded in the presence of the RTILs.36 Recently, Maroncelli and co-workers carried out a more detailed study on the photophysics of CVL in non polar and polar aprotic solvents37 and also in RTILs.38 Margulis and co-workers also showed via computational studies that the thermodynamics and the kinetics of the intramolecular ET of CVL in [Pr3+][NTf2−] arises from two different states.39
With the intension of suppressing one charge transfer band (CTB) of the CVL molecule in the longer wavelength region, we executed our study in a biomimicking organised assembly. To the best of our knowledge, this is the first report of the prevention of the CTA → CTB transformation of the CVL molecule in the presence of a nano-confined biomimicking organised assembly. The dual emission behaviour of the CVL molecule produces a single emission band in the presence of RMs. In the present study, our main focus is to decipher how the microenvironment surrounding the CVL molecule changes the photophysical behaviour entrapped in RM environments.
2. Experimental section
CVL (Scheme 1) was procured from Sigma-Aldrich and recrystallised twice with acetone prior to use. Sodium dioctylsulfosuccinate (AOT) (Scheme 1) was procured from Sigma-Aldrich. AOT was dried under vacuum for more than 48 h using a process described in our earlier publication.40 The concentration of the surfactant was kept constant (as 0.1 M) for all the experiments. Reverse micelles were prepared by the addition of a calculated amount of the respective polar solvent in an isooctane/AOT mixture at different w values (where w = [polar solvent]/[AOT]). For the preparation of water reverse micelles, we used Millipore water. Isooctane (spectroscopic grade) was procured from Spectrochem, India. DMF (HPLC grade) and glycerol (ACS grade) were purchased from RANKEM, India. Ethylene glycol was purchased from CDH, India. The concentration of CVL was maintained as 5 × 10−6 M. In each experiment, we used freshly prepared solutions. All the experimental measurements were performed at 298 K. | ||
| Scheme 1 Schematic of the CVL molecule in two different states and NaAOT surfactant. | ||
The ground state absorption spectra were obtained using a Shimadzu UV-Vis spectrophotometer (Model: UV-2550). The fluorescence emission spectra were recorded using a Horiba Jobin Yvon spectrofluorometer (Model: Fluoromax-4P). During the steady state absorption and fluorescence experiments, the temperature was maintained at 298 K using a Jeiotech refrigerated bath circulator (Model: RW0525G). The fluorescence quantum yield of CVL was calculated from the following equation using quinine sulphate solution in 0.1 N H2SO4 (ΦR = 0.546) as a reference.41
 | (1) |
 | (2) |
Time-resolved fluorescence emission measurements were carried out by the picosecond time-correlated single-photon counting (TCSPC) technique (Model: LifeSpec-II, Edinburgh Instruments, U.K.) using a light-emitting diode (LED, λexi = 340 nm) and a picosecond diode laser (λexi = 375 nm). The instrument response function (IRF) of our setup using LED and a diode laser is ∼800 ps and ∼75 ps, respectively. The fluorescence signals were collected at magic angle (54.7°) polarization using a Hamamatsu MCP PMT (3809U) as a detector. The decays were deconvoluted using the F-900 decay analysis software. Using the same instrument, we measured fluorescence anisotropy decays r(t). For the anisotropy decays, the polarised fluorescence emission intensities at parallel (I‖) and perpendicular (I⊥) polarizations were collected following excitation by the vertically polarised light. We used motorised polarizers to collect the parallel and perpendicular decays. The anisotropy decay r(t) was calculated as follows:
 | (3) |
3. Results & discussion
3.1 Steady state absorption studies
The solubility of CVL in water is low (1 mg ml−1 at 295 K). In an aqueous medium, CVL exhibits a structureless absorption maximum at 363 nm. CVL, entrapped in the AOT/isooctane mixture (w = 0), shows a prominent absorption maximum at 342 nm along with a hyperchromic effect. Herein, the absorption maximum is blue shifted, compared to neat water (Fig. 1a). The higher absorbance value and the blue-shifted absorption spectra of the CVL molecule in the AOT/isooctane mixture suggest that electrostatic interactions may prevail between the CVL and the polar head groups of the AOT. With the gradual addition of water in the RMs, a gradual increase in the absorbance was observed. In different non-aqueous RMs, we observed blue shifted absorption spectra compared to the respective polar solvent medium (Table 1) accompanied by a hyperchromic effect. This result clearly suggests that the microenvironment of the fluorophore is modified in restricted environments. In all the RMs, we observed a hypsochromic shift of the absorption maxima of the CVL molecule compared to the respective polar solvents. | ||
| Fig. 1 The change in the absorption spectra of CVL in (a) water RM, (b) DMF RM, (c) EG RM and (d) glycerol RM with variation of respective ‘w’ values and in corresponding neat solvents. | ||
| Sr no | System | w | λabsmax (nm) | λemimax (nm) | ϕfa | knra (ns−1) | τ1 (ns) | a1 | τ2 (ns) | a2 | 〈τ〉a (ns) | χ2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Experimental error of ±5% 〈τ〉 = a1τ1 + a2τ2. | ||||||||||||
| 1 | CVL in AOT/isooctane mixture | 0 | 342 | 397 | 0.19 | 0.153 | 3.46 | 0.68 | 9.14 | 0.32 | 5.28 | 1.02 |
| 2 | CVL in water | — | 363 | 440 | 0.16 | 0.173 | 2.28 | 0.69 | 10.60 | 0.31 | 4.86 | 1.02 |
| 3 | CVL in water/AOT/isooctane RM | 1 | 342 | 395 | 0.16 | 0.206 | 2.75 | 0.54 | 5.63 | 0.46 | 4.07 | 1.03 |
| 3 | 342 | 388 | 0.11 | 0.245 | 1.45 | 0.29 | 4.52 | 0.71 | 3.63 | 0.94 | ||
| 5 | 342 | 387 | 0.09 | 0.270 | 1.22 | 0.27 | 4.15 | 0.73 | 3.36 | 0.94 | ||
| 10 | 342 | 387 | 0.09 | 0.273 | 1.20 | 0.29 | 4.20 | 0.71 | 3.33 | 0.98 | ||
| 30 | 342 | 387 | 0.08 | 0.279 | 1.18 | 0.28 | 4.12 | 0.72 | 3.30 | 1.04 | ||
| 4 | CVL in DMF | — | 358 | 438 | 0.001 | 0.602 | 0.97 | 0.89 | 7.28 | 0.11 | 1.66 | 1.12 |
| 5 | CVL in DMF/AOT/isooctane RM | 1 | 343 | 412 | 0.19 | 0.129 | 3.56 | 0.56 | 9.70 | 0.44 | 6.26 | 1.03 |
| 2 | 343 | 419 | 0.14 | 0.173 | 2.43 | 0.54 | 7.93 | 0.46 | 4.96 | 1.05 | ||
| 3 | 343 | 422 | 0.10 | 0.245 | 1.72 | 0.57 | 6.26 | 0.43 | 3.67 | 1.01 | ||
| 6 | CVL in EG | — | 363 | 390 | 0.008 | 0.635 | 1.61 | 0.86 | 12.22 | 0.14 | 3.10 | 1.34 |
| 7 | CVL in EG/AOT/isooctane RM | 1 | 343 | 396 | 0.16 | 0.218 | 2.90 | 0.70 | 6.04 | 0.30 | 3.84 | 1.04 |
| 2 | 343 | 393 | 0.11 | 0.275 | 1.33 | 0.31 | 4.10 | 0.69 | 3.24 | 0.97 | ||
| 8 | CVL in glycerol | — | 361 | 424 | 0.052 | 0.491 | 1.30 | 0.78 | 6.30 | 0.22 | 2.40 | 1.29 |
| 9 | CVL in glycerol/AOT/isooctane RM | 1 | 342 | 396 | 0.17 | 0.186 | 3.31 | 0.71 | 7.28 | 0.29 | 4.46 | 1.06 |
| 2 | 342 | 393 | 0.14 | 0.203 | 3.17 | 0.68 | 6.51 | 0.32 | 4.24 | 1.02 | ||
| 3.5 | 342 | 390 | 0.12 | 0.218 | 2.65 | 0.49 | 5.38 | 0.51 | 4.04 | 1.02 | ||
The absorption maximum does not show any alteration with a gradual increase in w. This result can be ascribed to the lower ground state dipole moment (μg = 5.5 D) of the CVL molecule.30 Although in RMs, the gradual swelling of the polar solvent molecule changes the polarity of the medium, the differences in the polarity are not sufficient to cause a change in the ground state absorption maxima of CVL entrapped in RMs. Samanta and coworkers36 also showed that small differences in the polarity of RTILs are not sufficient to cause a change in the ground state absorption maximum of CVL. The polarity of the neat solvent molecules is greater than the polarity of the solvent entrapped in the RM cores. Therefore, CVL molecule entrapped in a RM exhibits a blue shifted absorption spectrum compared to that in the neat solvent. Fig. 1 shows the modification of the absorption spectra of a CVL molecule in different RMs and their respective neat solvents.
3.2 Steady state fluorescence emission studies
We used two different excitation wavelengths to monitor the excitation wavelength dependent fluorescence emission properties of CVL in RMs. In neat water, CVL exhibits an emission maximum at 440 nm but in the AOT/isooctane mixture, the emission maximum was observed at 397 nm (λexi = 340 nm). The hypsochromic shift of the emission maximum indicates that the microenvironment of the probe molecule is being modulated. In protic solvents, CVL forms cationic species followed by the solvent-aided heterocyclic dissociation of the C–O bond in the lactone ring.30 This photodissociation process of CVL in protic solvents takes place rapidly and the diabatic formation of the zwitterionic species is stabilised mainly via hydrogen bonding with the solvent molecule.30 In the presence of all neat solvents, we found a single emission maximum of CVL, except in neat DMF (herein, we observed a shoulder at the red end). Hydrogen bond donating solvent molecules (protic solvents) cause the retardation of electron transfer from one of the dimethyl aniline groups to the 6-DMAPd unit and therefore, instead of dual emission, we found a single emission band.30 In the case of highly viscous solvents, the solvent reorganisation energy is low, thereby the CTA → CTB transformation is prohibited and as a result, a single emission band is observed.36 The highly viscous solvents entrapped in the RM cores enhance the fluorescence quantum yield and retard the non-radiative deactivation process. On the gradual addition of viscous solvent to the respective RMs, the fluorescence quantum yield gradually decreases along with an increase in the non-radiative deactivation process. The dual emission band of CVL in a polar aprotic solvent (DMF) is attributed to the charge transfer species (CTA and CTB) of both the emitting states.30,37In isooctane, the CVL molecule exhibited an absorption maximum at 267 nm along with more absorption bands in the 200–450 nm region. Karpiuk also observed similar types of absorption spectral features of the CVL molecule in non-polar solvents such as hexane.30 They also explained the appearance of more absorption bands in the presence of non-polar solvents. We also found that the CVL molecule is soluble in nonpolar solvents like isooctane and hexane. In neat water, the solubility of the CVL molecule is very weak. CVL molecules entrapped in RMs exhibit an absorption maximum at 342 nm (shown in Fig. S1†). This clearly suggests that the dye molecule faces a different environment inside the RMs compared to in the isooctane medium. Moreover, in the presence of isooctane, the emission maximum of CVL appeared at 384 nm. Karpiuk found the emission maximum of CVL in hexane in the same region.30 The emission maximum of the CVL molecule inside the RMs was found to be red shifted compared to that in neat isooctane (Fig. S1a†). This result also clearly indicates that the probe molecule faces different microenvironments compared to that in neat isooctane. Moreover, in the presence of neat isooctane, excitation wavelength dependent emission spectral behaviour was also observed. This may be due to the contribution of impurities present in the dye molecule.44 From a plot of the fluorescence intensity against [AOT] (shown in Fig. S1b†), we calculated the partition coefficients of the CVL molecule at different ‘w’ values. The partition coefficient of the CVL molecule between the micellar surfaces compared to that in isooctane is high, indicating that the dye molecule resides at the micellar surface. The partition coefficients of the CVL molecule in an AOT/isooctane mixture and in water RMs (w = 5) are listed in Table S1.†
The water molecules entrapped in the bound layer of AOT surfactants exhibit different physical behaviour compared to neat water, and the ‘bound’ water molecules have a propensity to form strong hydrogen bonds with the head groups of the AOT surfactants. Thereby, the ‘bound’ water molecules entrapped in the RM cores exhibit a comparative drop in polarity, mobility, and intra-solvent hydrogen bonding networks and a comparatively higher viscosity.3–7,12,15,40,43 With the gradual swelling of the water pool in the aqueous RM cores, we found a steady decrease of the fluorescence intensity along with a hypsochromic shift of the emission spectra (Fig. 2a) when λexi = 340 nm. This hypsochromic shift of the emission spectra of the CVL molecule looks quite astonishing.
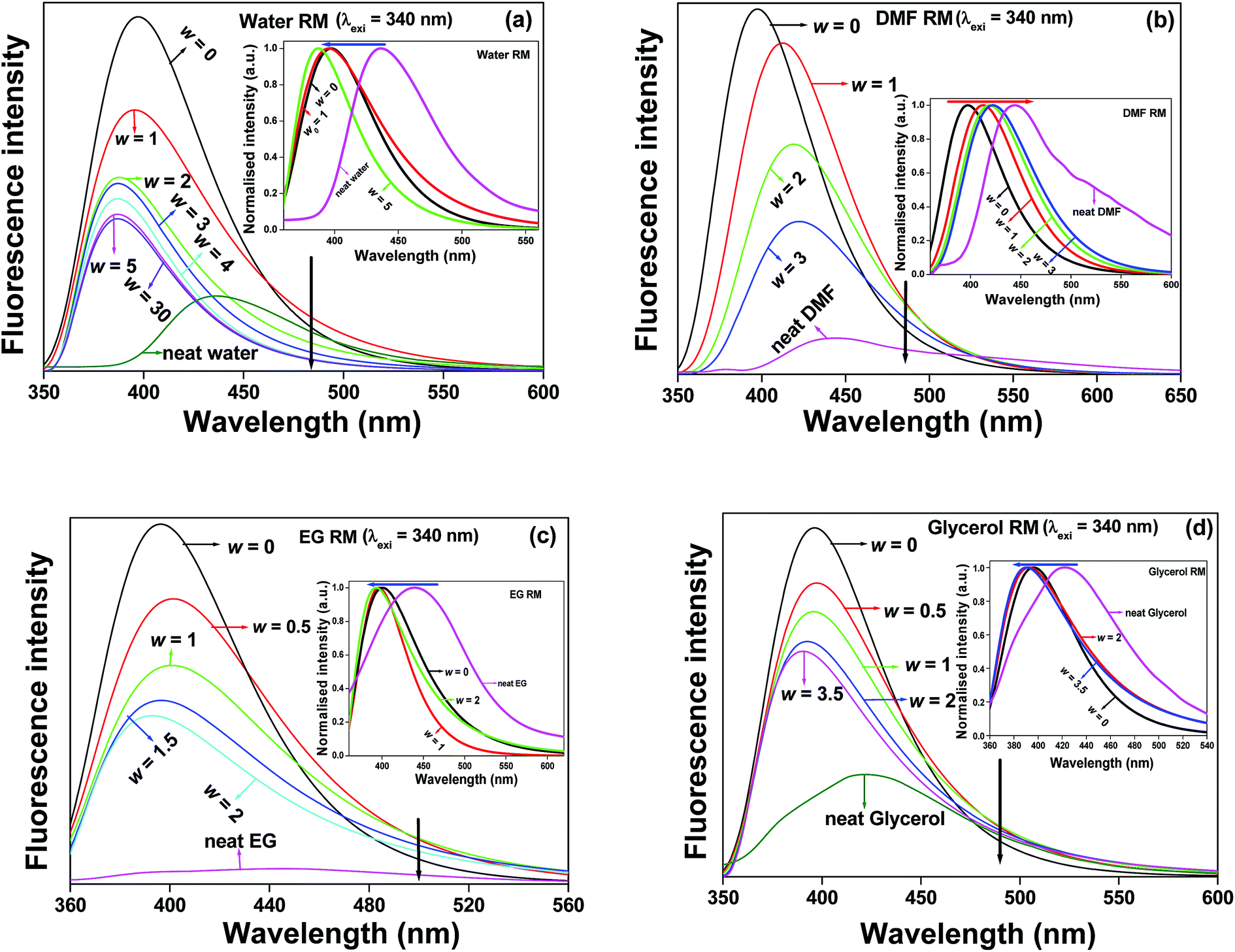 | ||
| Fig. 2 The emission spectral profiles of CVL (λexi = 340 nm) in (a) water RM, (b) DMF RM, (c) EG RM and (d) glycerol RM with variation of the respective ‘w’ values and the corresponding neat solvents. Insets of the respective figures show the changes in the emission spectral positions. | ||
When λexi = 340 nm, it is expected that CVL molecules residing at less polar regions are preferentially excited.45 Sengupta and co-workers46 also reported a hypsochromic shift in the emission spectra of the prodan molecule in water RMs with the gradual swelling of the water pool. They also inferred that the hypsochromic shift of the dye molecule is due to the presence of a probe molecule in the interfacial region or in the tail domain, thereby resulting in a highly non-polar environment. In our case, we observe a hypsochromic shift in the emission spectra with the gradual swelling of the water pool. Therefore, we can expect that some CVL molecules reside at the interfacial region. Furthermore, our proposition is in accordance with our previous study on the emission spectra of CVL in isooctane, wherein the emission maximum appears at 384 nm. Karpiuk found the emission maximum of CVL in hexane in the same region.30 Moreover, we found a high partition coefficient for the CVL molecule at a higher ‘w’ value, which clearly demonstrates that the probe molecule resides at the interfacial region of the micellar periphery. Similar types of hypsochromic effects on the emission spectra of CVL were also found with the gradual swelling of the solvent pool inside the RM cores by EG and glycerol (Fig. 2c and d), wherein the emission band undergoes a hypsochromic shift with gradually increasing ‘w’ values. From a closer scrutiny of the structure of the fluorophore, it is assumed that the less polar moieties of the dye molecules are preferentially excited when λexi = 340 nm. As a result, the dye molecules face a less polar region and have a tendency to avoid the protic solvent pool entrapped in the RM cores. Amararene et al.47 also showed by small angle X-ray scattering (SAXS) measurements that the aqueous core of the RMs in isooctane gradually increases from ‘w’ = 3 to 30. Therefore, it may be expected that on the gradual swelling of the water pool inside the RMs, there must be a change of the photophysical behaviour of the CVL molecule in water RMs. However, we found that at λexi = 340 nm and the gradual swelling of the water pool inside the RMs from ‘w’ = 5 to 30, the change of the fluorescence intensity, the fluorescence quantum yield and average lifetimes, along with their lifetime components and relative amplitudes are insignificant. It appears that the location of the dye molecule does not change with an increase in the size of the aqueous core of the RMs. The dye molecule is very weakly soluble in aqueous media. Hence, at higher ‘w’ values, the dye molecule has a propensity to reside at the less polar interfacial region and thereby exhibits no significant change in the spectral or the photophysical behaviour.
In the case of DMF RMs, we observed that the emission maximum of the CVL molecule is gradually red-shifted with the gradual swelling of the RMs pool (Fig. 2b). This implies that the dye molecule migrates towards the centre of the solvent pool from the interfacial region. The discernible modification of the emission spectra of CVL in DMF RMs suggests that the dye molecule undergoes charge dipole type interactions between the negatively charged sulphonate head groups of AOT and the highly charge-separated CVL because of the greater solvation of the Na+ counter cation by the polar coordinating solvent, DMF, thereby disrupting the weak intermolecular interaction between the DMF molecules.15,16,23,24 Karpiuk30 reported that in polar aprotic DMF solvents, CVL exhibits two bands (CTA and CTB). The CTA band originates from the polar excited state within the 6-DMAPd unit, whereas the CTB band originates from the highly polar excited state via the electron transfer (ET) from dimethylaniline (DMA) to the 6-DMAPd moieties. The polarity of CTA is higher than that of CTB in neat DMF. The most astonishing phenomenon associated with the CVL molecule in DMF RMs is that due to the decrement of polarity, we obtained a single emission band (CTA). The absence of the CTB band clearly indicates that the electron transfer (ET) mechanism is retarded inside the DMF RMs. Moreover, the species corresponding to λexi = 340 nm faces a comparatively less polar environment. In all the cases, we found that the fluorescence emission maxima of CVL entrapped in the respective RM cores undergo hypsochromic shifts compared to those of CVL in the respective neat solvents.
The photophysics of the emission spectra of CVL excited at a higher excitation wavelength (λexi = 375 nm) reflect different scenarios. The micropolarity, the microviscosity and the extent of hydrogen bonding of CVL inside the solvent pool of the different RMs causes the modulation of the fluorescence emission behavior. With the gradual trapping of the polar solvent inside the RM cores, the emission band of CVL undergoes a bathochromic shift of the emission maxima along with a decrease in the fluorescence intensity (Fig. S2†). This indicates that the fluorophore migrates towards the solvent pool of the RMs from the interfacial region with an increase in the w value.40,43 In the case of water RMs, with a gradual increase in the ‘w’ value, the fluorescence intensity along with different photophysical parameters significantly decreases. The emission spectral position of the dye molecule also undergoes a significant bathochromic shift with increasing ‘w’ values up to ‘w’ = 5. However, there is no significant change in the fluorescence intensity or the average lifetime values along with the lifetime components and their relative population beyond ‘w’ = 5 (as shown in Fig. S2a and Table S2†). Astonishingly, at λexi = 375 nm, the emission maximum of CVL beyond ‘w’ = 5 undergoes a hypsochromic shift (Table S2†). This may be due to the fact that the aqueous solubility of the CVL molecule is very weak. Hence, at higher water contents of the aqueous core of the RMs, the dye molecule has a propensity to locate at the interfacial region of the solvent pool. The photophysical parameters of CVL in different neat solvents and their respective RMs (at different ‘w’ values) at two different excitation wavelengths (λexi = 340 nm and 375 nm) are tabulated in Table 1 and Table S2.† From Table S2,† it was observed that the microenvironment of the dye molecule entrapped in a RM is completely different from that in the respective neat solvent. The probe molecule faces a comparatively less polar environment inside the solvent pool of the respective RM compared to that in the respective neat solvent. With the gradual swelling of the RMs, the fluorescence quantum yields decrease. As a result, the non-radiative deactivation process becomes very fast and causes CVL molecules to become less fluorescent in nature.
In order to comprehend the excitation wavelength dependency of steady state emission spectra, we carried out steady state emission studies of CVL at different excitation wavelengths. Astonishingly, we noticed insignificant shifts of the emission maxima of CVL in neat solvents like water and DMF with changes in the excitation wavelengths. However, appreciable shifts in the emission maxima were observed in the case of viscous solvents like EG and glycerol (Fig. S3†), which show the presence of specific regions of order as described by McDuffie and Litovitz.48 A CVL molecule entrapped in isooctane/AOT mixtures and in different RMs also exhibits excitation wavelength dependent emission behaviour (Fig. S4†). The emission spectral shift of the CVL molecule was tabulated in Table S3† by varying the excitation wavelengths from 340 to 405 nm. Previously, Maroncelli and co-workers also reported the excitation wavelength dependence of electron transfer (ET) between two polar excited states of CVL in [Pr3+][NTf2−].38 However, the same group also concluded that contributions from the impurities present in the sample may cause the excitation wavelength dependency behaviour.44 Therefore, the presence of impurities may also contribute to the dependence of the emission wavelength of CVL on the excitation energy.
In the case of water, EG and glycerol RMs, we noticed that on gradual swelling of the RM cores, the hypsochromic shift of the emission band of CVL is observed when the excitation wavelength is varied from λexi = 340 nm to 360 nm, while the opposite spectral features are noticed when λexi is varied from 375 nm to 405 nm (Fig. S5†). The emission band undergoes a bathochromic shift with a gradual increase in the ‘w’ values at higher excitation wavelengths (λexi = 375 nm to 405 nm). This opposing emission behaviour of CVL entrapped in RMs at different excitation wavelengths may be assumed to be due to the distribution of the fluorophores in different locations of the RMs, resulting in different types of solute–solvent interactions. We may also assume that the effects of micropolarity and microviscosity act in combination at different excitation wavelengths, thereby resulting in different emission behaviour by the CVL molecule in the aforesaid systems. However, in the case of DMF RMs, we noticed that at different excitation wavelengths (λexi = 340 nm to 405 nm), a bathochromic emission shift for the CVL molecule is observed with a gradual increase of the ‘w’ value. The addition of the non-hydrogen-bond-donating solvent, DMF, causes the solvation of the Na+ counter ion inside the pool of the RM and hence the structure of DMF is highly disrupted.23,24 Therefore, the unfunctionalized polar sulfonate head groups of the AOT molecule undergo charge-dipole type interactions with the charge separated CVL molecule and exhibit a bathochromic emission shift with a gradual increase in the ‘w’ value. As reported, the CTA band originates from the polar excited state, which is localised within the 6-DMAPd units and its dipole moment is 10.7 D.38 Henceforth, it may be assumed that increasing the micropolarity causes the migration of the CVL molecule towards the RM core from the interfacial region. As the excitation wavelength is monitored towards the longer wavelength region, the species corresponding to the more polar region becomes excited and hence exhibits a bathochromic emission band. Moreover, the CVL molecule entrapped in the restricted environment exhibits a single emission band (CTA) instead of a dual emission band (CTA and CTB) (Scheme 2). The excitation wavelength dependent fluorescence emission behavior also reflects the change of full width half maxima (FWHM) of emission spectra of CVL at different ‘w’ values in the studied RMs at different excitation wavelengths (Fig. S6†).
 | ||
| Scheme 2 Schematic of the plausible interaction of the solvent sequestered with the CVL (1CTA) species present in the core of RMs containing polar protic solvents and absence of 1CTB species at λexi = 375 nm. | ||
3.3 Time resolved emission studies
In order to explore the excited state dynamics and the probable location of the fluorophore entrapped in different RMs, we investigated the time resolved fluorescence emission properties of CVL by varying the excitation wavelengths (λexi = 340 nm and 375 nm) at different ‘w’ values. The fluorescence lifetime decays are fitted using the multiexponential function. The fluorescence quantum yield values (Fig. S7†) and the average decay times gradually decrease with a gradually increasing ‘w’ value at two different excitation wavelengths (Fig. S8†). The non-radiative decay rate constants (knr) for CVL at two different excitation wavelengths are different and these values gradually increase with a gradual increase in the ‘w’ value in the RMs (Fig. S9†). From a closer scrutiny of average lifetime values, non-radiative decay rate constants and the fluorescence quantum yield values of CVL at two different excitation wavelengths, it is clear that the CVL molecule showed excitation wavelength dependency. The fluorescence lifetime components (τi) and the respective populations (ai) (Table 1 and Table S2†) of CVL at two different excitation wavelengths in neat solvents, AOT/isooctane mixture and in the respective RMs have significant differences this signifying that the microenvironments of CVL are modulated. Fig. 3 displays the fluorescence decay time of CVL in various neat solvents and in the respective RMs when λexi = 340 nm. In all the RMs, we fitted the lifetime decays using a biexponential function comprising both the fast (τ1) and the slow (τ2) components. In the case of water RMs, with gradual swelling of the water pool inside the RMs, both the components gradually decrease in a systematic pattern. On a closer scrutiny of the lifetime components and their relative populations in water RMs, we found that with a gradual increase in the ‘w’ value, the relative amplitude (a1) of the fast component (τ1) gradually decreases, while the relative amplitude (a2) of the slow component (τ2) gradually increases, causing a decrease in the average lifetime value of CVL. Such an observation clearly indicates that the probe molecule faces a different polarity in the RMs. It can be noted that in neat isooctane, we found that CVL exhibits a single exponential lifetime component of 4.58 ns. In water RMs, with a gradual increase in the ‘w’ value, a similar type of long component has been noticed, with a gradual increase of its relative population. Therefore, it may be assumed that the species corresponding to λexi = 340 nm have a tendency to reside at the less polar region. For that reason, we noticed the hypsochromic emission band with a gradual increase in the ‘w’ value. Similar observations were made in the case of EG and glycerol RMs. The gradual entrapping of viscous solvents by the aforesaid RMs caused a decrease in both the fast component (τ1) and the slow component (τ2). The relative amplitude of the fast component (a1) significantly decreases at higher ‘w’ values, whereas the relative amplitude of the slow component (a2) significantly increases. Such observations can be rationalized by considering the fact that the probe molecules located at the interfacial regions of the AOT/isooctane mixtures have a propensity to migrate towards the comparatively less polar microenvironments in the case of water, EG and glycerol RMs (at λexi = 340 nm). | ||
| Fig. 3 The fluorescence emission decay profiles of CVL (λexi = 340 nm) in (a) water RM, (b) DMF RM, (c) EG RM and (d) glycerol RM with variation of respective ‘w’ values and the corresponding neat solvents. | ||
In the case of the DMF RMs, the scenario is completely different. At λexi = 340 nm, CVL molecules migrate towards the DMF solvent pool through charge-dipole interaction and as a result, with increasing ‘w’ value, both the components decrease according to a regular pattern but the relative populations of both the components do not significantly change. Moreover, we observed that the magnitude of the slow component of CVL at a higher ‘w’ value is different to the component observed in isooctane (4.58 ns). This indicates that CVL in DMF RMs have less probability to reside in the less polar environments, and the charge-dipole interaction takes place in the RMs pool. Moreover, it can be noted that in DMF RMs, a bathochromic shift in emission band for CVL has been observed with a gradual increase in ‘w’ (Table 1).
The probe molecule located in a comparatively more polar region (λexi = 375 nm) exhibited a different scenario. In all the RMs, the lifetime decays are fitted using a triexponential function. In the case of λexi = 340 nm, we used a light emitting diode (LED) and the instrument response function (IRF) is ∼800 ps, whereas at λexi = 375 nm, a picosecond diode laser is used and the value of IRF is ∼75 ps. Therefore, at λexi = 340 nm, we are unable to get the fast component value. At both the excitation wavelengths, the different component may be attributed to the interaction between the dye molecule and the solvent pool inside the RMs and in the interfacial region. The average fluorescence lifetime values (Table S2 and Fig. S10†) and their lifetime components gradually decrease as ‘w’ increases. This clearly suggests that on increasing ‘w’ values, the micropolarity of the solvent pool inside the RM core gradually increases, while the microviscosity of the medium gradually decreases, which leads to a decrease in the excited state lifetime and an increase in the non-radiative deactivation pathway. Moreover, the probe molecule suffers a less restricted environment with a gradual increase in ‘w’.
This clearly suggests that when λexi = 375 nm, the CVL molecules migrating towards the more polar region of the solvent pool of the RMs are preferentially excited. When λexi = 375 nm, the long component value (τ3) and its relative population (a3) gradually decreases (Table S2†) on increasing the value of ‘w’ in all the studied RMs. Astonishingly, we observed that compared to λexi = 340 nm, an opposing tendency of the relative population of the long component of CVL entrapped in water, EG and glycerol RMs is observed when λexi = 375 nm. When λexi = 375 nm, a single exponential decay for CVL was found in isooctane and the lifetime value is 4.58 ns. In this regard, it is pertinent to mention that when λexi = 375 nm, the tendency of the CVL molecule is to reside in the more polar region of RMs.
We observed that CVL molecules entrapped in both aqueous and non-aqueous RMs exhibit emission wavelength dependent emission behaviour. We recorded emission decays of probe molecules at the emission maxima, the far red end and the blue end of the emission spectra and found considerable differences in the lifetime decays. The emission wavelength dependent lifetime decays of CVL with different RMs at two different excitation wavelengths are shown in Fig. 4 and Fig. S11–S13.† The excitation and the emission wavelength dependency of the CVL molecule entrapped in the RM assembly are due to the presence of an ensemble of molecules in the ground state and the inherent heterogeneity of the environments and hence energies. The emission wavelength dependent spectral differences arise primarily due to the distribution of the probe molecule into different microheterogeneous regions of solvent, causing different rates for the excited state spectral relaxation process.
 | ||
| Fig. 4 The emission wavelength dependent fluorescence emission decays of CVL in glycerol RMs at two different excitation wavelengths (a) λexi = 340 nm and (b) λexi = 375 nm. | ||
3.4 Time resolved anisotropy studies
Time-resolved fluorescence anisotropy measurements are a sensitive tool for gathering knowledge about the rotational relaxation of probe molecules and the rigidity of environments. When the fluorophores in confined media show multiple lifetime components and exhibit dynamic behaviour, then one has to use anisotropy decays and analyse them for a meaningful physical explanation. Anisotropy rotation gleans the motional restriction conferred on surrounding micellar media on the fluorophore, which can indicate the probable location of the molecule in confined media. The anisotropy decays of CVL molecules entrapped in RMs are biexponential in nature (λexi = 375 nm) and can be constructed as follows:
r(t) = r0[β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τslow) + (1 − β)exp(−t/τfast)] exp(−t/τslow) + (1 − β)exp(−t/τfast)]
| (4) |
| 〈τrot〉 = βτslow + (1 − β)τfast | (5) |
In neat water, the anisotropy decay of CVL is fitted using a single exponential function and the rotational relaxation time is 120 ps. The rotational relaxation time significantly increases when CVL molecules are entrapped in aqueous RMs and the anisotropy decay is biexponential in nature. This infers that the probe molecule is facing a more restricted environment. On increasing ‘w’ values, the fluorophore experiences a less confined environment and the rotational relaxation time gradually decreases. The rotation process gradually becomes fast on gradual swelling of the water inside the RM pool. Exploration of Table 2 indicates that in the case of the water RMs, at higher ‘w’ values, both the fast component (τfast) and the slow component (τslow) gradually decrease along with their relative amplitudes. In the case of neat DMF, single exponential anisotropy decay is found and the rotational relaxation time is 300 ps. In DMF RMs, the rotational process for CVL becomes significantly slower due to the restricted environments faced by the probe molecule. The rotational relaxation time significantly increases and it is observed that on increasing the value of ‘w’, the rotational relaxation time gradually decreases, which signifies that the rotational dynamics become gradually fast and the microenvironment becomes less confined. Similar observations were made in our earlier report.22 The anisotropy decays of CVL in neat solvents and in their respective RMs are shown in Fig. 5 and S14.†
| System | w | r0 | τ1 (ns) | a1 | τ2 (ns) | a2 | 〈 τ〉rota (ns) | χ2 |
|---|---|---|---|---|---|---|---|---|
| a Experimental error of ±5% 〈τ〉rot = a1τ1 + a2τ2 + a3τ3. | ||||||||
| CVL in AOT/isooctane mixture | 0 | 0.33 | 0.51 | 0.26 | 2.18 | 0.74 | 1.75 | 1.10 |
| CVL in water | — | 0.30 | 0.12 | 1.00 | 0.12 | 1.21 | ||
| CVL in isooctane/AOT/water RM | 1 | 0.26 | 0.87 | 0.42 | 2.98 | 0.58 | 2.09 | 1.04 |
| 5 | 0.35 | 0.36 | 0.60 | 2.88 | 0.40 | 1.37 | 0.90 | |
| 10 | 0.32 | 0.32 | 0.70 | 1.71 | 0.30 | 0.74 | 1.02 | |
| CVL in DMF | — | 0.31 | 0.30 | 1.00 | 0.30 | 1.06 | ||
| CVL in isooctane/AOT/DMF RM | 1 | 0.22 | 0.39 | 0.32 | 1.98 | 0.68 | 1.47 | 1.12 |
| 3 | 0.24 | 0.32 | 0.57 | 2.13 | 0.43 | 1.10 | 1.13 | |
| CVL in EG | — | 0.38 | 2.15 | 1.00 | 2.15 | 1.04 | ||
| CVL in isooctane/AOT/EG RM | 1 | 0.29 | 0.91 | 0.46 | 4.92 | 0.54 | 3.08 | 1.16 |
| 2 | 0.30 | 0.84 | 0.43 | 6.18 | 0.57 | 3.88 | 1.00 | |
| CVL in glycerol | — | Hindered rotation | ||||||
| CVL in isooctane/AOT/Glycerol RM | 1 | 0.29 | 1.93 | 0.39 | 8.10 | 0.61 | 5.69 | 1.09 |
| 2 | 0.31 | 1.82 | 0.29 | 15.03 | 0.71 | 11.20 | 1.08 | |
| 3.5 | 0.30 | 2.72 | 0.24 | 26.73 | 0.76 | 20.97 | 1.10 | |
 | ||
| Fig. 5 The time-resolved fluorescence anisotropy decays of CVL in (a) water RM and (b) glycerol RM, when λexi = 375 nm. | ||
In the case of neat glycerol, the rotational relaxation of CVL is completely hindered and we are unable to detect the rotational relaxation time of CVL due to the highly restricted environments. A similar result was reported earlier.49 CVL entrapped in glycerol RMs shows comparatively fast rotational relaxation behaviour as we observe that the rotational relaxation time increases from 5.7 ns at w = 1 to 20 ns at w = 3.5. This is quite an astonishing feature as we observe the opposite effect of microviscosity on the rotational relaxation behaviour of CVL. In neat glycerol, the specific solute–solvent interaction is mainly prominent in among the probe, CVL, and glycerol thereby creating a highly viscous microenvironment by means of strong friction between the rotating particle and the solvent.
In the case of glycerol RMs, the main specific solute–solvent interaction takes place between the sulphonate head group of AOT and glycerol. Henceforth, the specific solute solvent type interaction between the probe and trapped glycerol is weaker than that in neat solvent. This reduces the friction between the rotating dye and the surrounding trapped solvent molecule. In summary, in the case of neat glycerol, the rotational relaxation time is too long to measure. However, for the dye molecule entrapped in the glycerol/AOT/isooctane RMs, we are able to measure the rotational relaxation time. The rotation time for CVL in glycerol is too long to measure. At higher ‘w’ values, the rotational relaxation time gradually increases and becomes longer in isooctane/AOT/Glycerol RM.
The rotational relaxation time of CVL in neat EG is 2.15 ns and the anisotropy decay is also single exponential in nature. The rotational relaxation time has been found to increase when CVL molecules are entrapped in the EG RMs. The dye molecule in the confined media causes the rotational process to be very slow. The most interesting observation is that on increasing the value of ‘w’, the rotational relaxation time of CVL gradually increases. In the case of the glycerol RMs, we found a similar trend. Such anomalous results for anisotropy decays of EG and glycerol based RMs appear contradictory. To explain such anomalous behaviour, we have to consider the effects of hydrodynamic friction and dielectric friction. It is assumed that both the types of friction couple together to produce such increases in rotational relaxation time with a gradual increase in ‘w’ value. Similar enhancements in rotational relaxation time were observed in EG and glycerol RMs in earlier reports.22,23 Laia and Costa,24 followed by Ferreira and Costa50 reported that on increasing the ‘w’ values of the RMs, the fluorescence anisotropy values gradually increased.
4. Conclusion
In summary, we investigated the photophysical properties of CVL in aqueous and non-aqueous RMs. The most remarkable observation is that in a polar aprotic solvent (DMF), the fluorophore (CVL) exhibits dual emission behavior (CTA and CTB states of CVL), whereas CVL entrapped in DMF RMs displays a single emission band (CTA). In all the studied RMs, we observed that the CTA → CTB transformation is retarded and a single emission band is found. The CTB band of CVL disappears in protic (H-bond donating) and viscous solvents. In water, EG and glycerol RMs, a hypsochromic shift of the emission band is observed on the gradual swelling of the RMs (at λexi = 340 nm). This may indicate that the CVL molecule is preferentially located at a less polar region. In DMF RMs, the bathochromic shift of the emission band on gradually increasing ‘w’ may indicate that the CVL molecule migrates from the interfacial region to the core of the RM via a charge-dipole and a specific solute–solvent interaction. When λexi = 375 nm, a bathochromic shift of the emission band for CVL is observed on a gradual increase in ‘w’. This may demonstrate that some probe molecules have migrated to the more polar region of the solvent pool of the respective RMs. Therefore, at different excitation wavelengths (λexi = 340 nm and 375 nm), CVL molecule entrapped in the restricted environment exhibits different photophysical behaviour in water, EG and glycerol RMs. CVL molecules also exhibit emission wavelength dependency in the confined media. Such observations arise mainly due to the distribution of the probe molecule into different microheterogeneous segments, resulting in different rates for the excited state spectral relaxation process. The rotational relaxation time of CVL is increased in the RMs compared to that in the respective neat solvents excepting neat glycerol. The rotational relaxation time gradually decreases on increasing ‘w’ in the case of water and DMF RMs, whereas it gradually increases in EG and glycerol RMs. It may be assumed that in the case of viscous solvent molecules entrapped in the RMs, the hydrodynamic friction and the dielectric friction couple together, thereby enhancing the rotational relaxation time.Acknowledgements
All the authors are thankful to the Indian Institute of Technology Patna (IIT Patna), India, for the research facilities. B.M. and S.A.A are thankful to IIT Patna, for research fellowships. A.C. is thankful to CSIR, New Delhi, for research fellowships.References
- Structure and Reactivity of Reverse Micelles, ed. M. P. Pileni, Elsevier, New York, 1989 Search PubMed.
- P. L. Luisi, M. Giomini, M. P. Pileni and B. H. Robinson, Biochim. Biophys. Acta, 1988, 947, 209–246 CrossRef CAS.
- Photochemistry in Microheterogenous Systems, ed. K. Kalyanasundram, Academic Press, New York, 1987 Search PubMed.
- Reverse Micelles: Bilogical and Technological Relevance of Amphiphilic Structure in Apolar Media, ed. P. L. Luisi and B. E. Straube, Springer, New York, 1999 Search PubMed.
- N. Nandi, K. Bhattacharyya and B. Bagchi, Chem. Rev., 2000, 100, 2013–2046 CrossRef CAS PubMed.
- D. C. Crans and N. E. Levinger, Acc. Chem. Res., 2012, 45, 1637–1645 CrossRef CAS PubMed.
- N. E. Levinger and L. A. Swafford, Annu. Rev. Phys. Chem., 2009, 60, 385–406 CrossRef CAS PubMed.
- Membrane Mimetic Chemistry: Characterizations and Applications of Micelles, Microemulsions, Monolayers, Bilayers, Vesicles, Host-Guest Systems, and Polyions, ed. J. H. Fendler, Wiley-Interscience, New York, 1982 Search PubMed.
- M. P. Pileni, J. Phys. Chem., 1993, 97, 6961–6973 CrossRef CAS.
- E. P. Melo, M. R. Aires-Barros and J. M. Cabral, Biotechnol. Annu. Rev., 2001, 7, 87–129 CAS.
- J. P. Wilcoxon, R. L. Williamson and R. Baughman, J. Chem. Phys., 1993, 98, 9933–9950 CrossRef CAS PubMed.
- M. Wong, J. K. Thomas and T. Nowak, J. Am. Chem. Soc., 1977, 99, 4730–4736 CrossRef CAS.
- F. Moyano, E. Setien, J. J. Silber and N. M. Correa, Langmuir, 2013, 29, 8245–8254 CrossRef CAS PubMed.
- L. C. Lee and Y. Zhao, J. Am. Chem. Soc., 2014, 136, 5579–5582 CrossRef CAS PubMed.
- N. M. Correa, J. J. Silber, R. E. Riter and N. E. Levinger, Chem. Rev., 2012, 112, 4569–4602 CrossRef CAS PubMed.
- R. D. Falcone, J. J. Silber and N. M. Correa, Phys. Chem. Chem. Phys., 2009, 11, 11096–11100 RSC.
- I. Danielsson and B. Lindman, Colloids Surf., 1981, 3, 391–392 CrossRef CAS.
- T. K. De and A. Maitra, Adv. Colloid Interface Sci., 1995, 59, 95–193 CrossRef CAS.
- S. P. Moulik and B. K Paul, Adv. Colloid Interface Sci., 1998, 78, 99–195 CrossRef CAS.
- A. M. Dokter, S. Woutersen and H. J. Bakker, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15355–15358 CrossRef CAS PubMed.
- H. Shirota and H. Segawa, Langmuir, 2004, 20, 329–335 CrossRef CAS.
- B. Maity, A. Chatterjee and D. Seth, Chem. Phys. Lett., 2013, 565, 108–115 CrossRef CAS PubMed.
- A. Chatterjee and D. Seth, Photochem. Photobiol. Sci., 2013, 12, 369–383 CAS.
- C. A. T. Laia and S. M. B. Costa, Langmuir, 2002, 18, 1494–1504 CrossRef CAS.
- D. G. Hayes and E. Gulari, Langmuir, 1995, 11, 4695–4702 CrossRef CAS.
- L. Mukherjee, N. Mitra, P. K. Bhattacharya and S. P. Moulik, Langmuir, 1995, 11, 2866–2871 CrossRef CAS.
- A. Chakraborty, D. Seth, P. Setua and N. Sarkar, J. Phys. Chem. B, 2006, 110, 5359–5366 CrossRef CAS PubMed.
- Z. R. Grabowski, Pure Appl. Chem., 1993, 65, 1751–1756 CrossRef CAS.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- J. Karpiuk, J. Phys. Chem. A, 2004, 108, 11183–11195 CrossRef CAS.
- M. A. White, J. Chem. Educ., 1998, 75, 1119–1120 CrossRef CAS.
- Y. Kaneko and D. C. Neckers, J. Phys. Chem. A, 1998, 102, 5356–5363 CrossRef CAS.
- Y. Ma, B. Zhu and K. Wu, Sol. Energy, 2001, 70, 417–422 CrossRef CAS.
- J. Karpiuk, Z. R. Grabowski and F. C. De Schryver, J. Phys. Chem., 1994, 98, 3247–3256 CrossRef CAS.
- J. Karpiuk, E. Karolak and J. Nowacki, Phys. Chem. Chem. Phys., 2010, 12, 8804–8809 RSC.
- K. Santhosh and A. Samanta, J. Phys. Chem. B, 2010, 114, 9195–9200 CrossRef CAS PubMed.
- X. Li and M. Maroncelli, J. Phys. Chem. B, 2011, 115, 3746–3754 CrossRef CAS PubMed.
- H. Jin, X. Li and M. Maroncelli, J. Phys. Chem. B, 2007, 111, 13473–13478 CrossRef CAS PubMed.
- H. V. R. Annapureddy and C. J. Margulis, J. Phys. Chem. B, 2009, 113, 12005–12012 CrossRef CAS PubMed.
- A. Chatterjee, B. Maity and D. Seth, Phys. Chem. Chem. Phys., 2013, 15, 1894–1906 RSC.
- G. A. Crosby and J. N. Demas, J. Phys. Chem., 1971, 75, 991–1024 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, 3rd edn, Springer, New York, 2006 Search PubMed.
- B. Maity, A. Chatterjee and D. Seth, RSC Adv., 2014, 4, 3461–3471 RSC.
- H. Jin, X. Li and M. Maroncelli, J. Phys. Chem. B, 2010, 114, 11370 CrossRef CAS.
- K. Bhattacharyya, Chem. Commun., 2008, 2848–2857 RSC.
- B. Sengupta, J. Guharay and P. K. Sengupta, Spectrochim. Acta, Part A, 2000, 56, 1433–1441 CrossRef.
- A. Amararene, M. Gindre, J.-Y. Le Huérou, W. Urbach, D. Valdez and M. Waks, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 61, 682–689 CrossRef CAS.
- G. E. McDuffie and T. A. Litovitz, J. Chem. Phys., 1962, 37, 1699–1705 CrossRef CAS PubMed.
- A. Chatterjee, B. Maity, S. A. A. Ahmed and D. Seth, J. Phys. Chem. B, 2014, 118, 12680–12691 CrossRef CAS PubMed.
- J. A. B. Ferreira and S. M. B. Costa, J. Phys. Chem. B, 2010, 114, 10417–10426 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The change of absorption and emission spectral features of CVL in isooctane and water RMs, steady state emission spectra, variation of FWHM with w value, excitation wavelength dependency of CVL in different RMs, variation of quantum yield, average fluorescence lifetime w value, the fluorescence emission decay profile and anisotropy decay of CVL in RMs and the photophysical parameters of CVL in RMs are all shown in the supporting information. See DOI: 10.1039/c5ra06309d |
| This journal is © The Royal Society of Chemistry 2015 |