
Free nitrous acid breaks down extracellular polymeric substances in waste activated sludge
Abstract
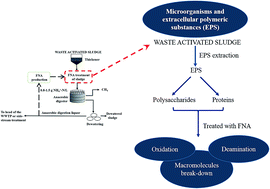
Free nitrous acid (FNA) has been demonstrated to be effective in enhancing the degradability of waste activated sludge (WAS). Considering that extracellular polymeric substances (EPS) are a major component in sludge flocs, the chemical breakdown of EPS components by FNA has been hypothesized to account for the improvement of sludge biodegradability in addition to enhanced cell lysis. EPS extracted from WAS was treated with FNA at 2.0 mg HNO2-N per L (260 mg NO2−-N per L and pH 5.5). The molecular weight distribution of EPS showed the breakdown of macromolecules into smaller molecules. The chemical structure analysis of EPS using Fourier transform infrared spectroscopy ascribed the breakdown to FNA-induced deamination of proteins, amino sugars and nucleic acids, implying that the main targets of FNA in EPS are protein-like substances. Particle size distribution analysis on the original WAS with the same FNA treatment revealed that FNA treatment of sludge significantly reduces the flocs sizes, which supported that FNA breaks down EPS in activated sludge flocs.
 Please wait while we load your content...
Please wait while we load your content...

