
A quest for stable 2,5-bis(halobora)cyclopentenylidene and its Si, Ge, Sn and Pb analogs at theoretical levels†
Abstract
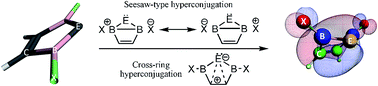
Replacing the two nitrogen atoms of Arduengo's N-heterocyclic carbenes (NHCs) with electron deficient boron atoms forms B-heterocyclic carbenes (BHCs) which may appear destabilizing at first glance. Yet, among the 40 optimized singlet (s) and triplet (t) BHCs and their Si, Ge, Sn and Pb homologues (BHËs), eight species are found that show higher stability than their corresponding NHËs for exhibiting wider singlet–triplet energy gaps (ΔEst), at B3LYP/TZ2P, as well as CBS-QB3 and G4MP2 ab initio levels. Moreover, triplet BHËs assume a planar geometry with a dihedral angle (D1) of about zero degrees. In contrast, their corresponding singlets show a high tendency for puckering with D1 ≅ 66°. The preference of the latter for puckered nonplanar geometries is evidenced by NBO calculations and visually through their frontier molecular orbitals. The main stabilizing interactions appear to be π- and σ-bond hyperconjugation across the ring. The resulting eight species that demonstrate higher stability are: 2,5-bis(iodobora)cyclopentensilylene, 2,5-bis(Z-bora)cyclopenten-germylene and -stannylene, for Z = Br and I; as well as 2,5-bis(Z2-bora)cyclopentenplumbylene, for Z2 = Cl, Br and I.
 Please wait while we load your content...
Please wait while we load your content...

