DOI:
10.1039/C5RA01416F
(Paper)
RSC Adv., 2015,
5, 43303-43311
Dithieno[3,2-a:2′,3′-c]phenazine-based chemical probe for anions: a spectroscopic study of binding†
Received
24th January 2015
, Accepted 27th April 2015
First published on 27th April 2015
Abstract
The synthesis of a new anion-responsive molecule N,N′-(2,5-bis(4-(tert-butyl)phenyl)dithieno[3,2-a:2′,3′-c]phenazine-9,10-diyl)bis(4-methylbenzenesulfonamide) (1) is reported. The sensitivities of the spectroscopic properties of 1 in the presence of various anions were examined using UV-vis absorption spectroscopy, fluorescence and 1H NMR titration experiments. Strong binding of 1 to carboxylate, cyanide, fluoride and dihydrogen phosphate anions results in an increase in quantum yield for emission of 1, and changes in its 1H NMR chemical shifts. A significant electrostatic interaction of the tetrabutylammonium cation with 1, upon strong binding with the counter anion, was also indicated by the chemical shifts observed in the 1H NMR titrations. Binding constants of 1 to anions are also calculated based on the binding isotherms derived from NMR and UV-vis titrations. DFT calculations show that the anion does not significantly impact the HOMO/LUMO levels (and subsequently the S0 → S1 transition), but rather changes the strength of the S0 → S2 transition, which accounts for the observed changes in the UV-vis spectra.
Introduction
The interest in designing optical probes to effectively detect anions stems from the anions' important role in biological1–6 and environmental systems.3,6 The ability to detect and monitor anions' presence, either qualitatively or quantitatively, is crucial for health and the environment. Halide anions are essential for the operation and regulation of enzymatic activities7–9 while the cyanide anion is extremely poisonous.10 Phosphate anions and their derivatives are fundamental components of biomolecules such as DNA and ATP; the phosphate anion component of these biomolecules is needed to determine their functions.11 Carboxylates, on the other hand, are present in amino acids, peptides and proteins.
The opto-electronic properties of benzodithiophenes have led these moieties to be compatible components for constructing molecular electronic devices. Fused benzodithiophene compounds are electrochemically active12,13 and they undergo electrochemical polymerization;14 extended systems are also fluorescent with high quantum yields.15,16 Substituted benzo[2,1-b:3,4-b′]dithiophene-4,5-dione have been recently reported in OFET devices13,17 while benzodithiophene-based compounds have been explored in OFET18–26 and OPV27,28 devices.
We29,30 and others31–35 have reported the use of sulfonamide groups to detect a wide range of anions. In this paper, we report on the preparation of the compound N,N′-(2,5-bis(4-(tert-butyl)phenyl)dithieno[3,2-a:2′,3′-c]phenazine-9,10-diyl)bis(4-methylbenzenesulfonamide) (1) (Scheme 1) and on the changes in its spectroscopic profile upon the addition of anions. The significant changes in the fluorescence profile of 1 upon the addition of benzoate, cyanide, or dihydrogen phosphate anions highlights its potential application as a fluorescent probe for these anions. Also, this combination of an anion-binding site with benzodithiophene fluorophore provides a supramolecular tool for tuning the electronic properties of benzodithiophenes.
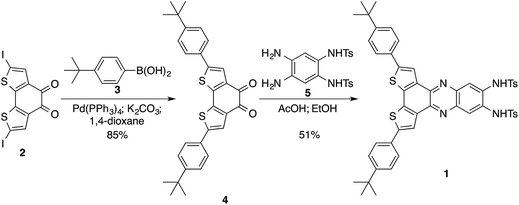 |
| | Scheme 1 Synthesis of optical probe 1. | |
Design and synthesis
In many cases fluorescent chemical probes are designed such that a binding site of the targeted analyte is linked to a fluorophore. If the linkage between the chromophore and the bridge group provides significant electronic coupling between the two parts, the binding event of the analyte can result in changes of electronic structure of the emissive part of the molecule, and hence changes of its absorption and emission properties. These changes in the optical properties are dependent on several factors: (i) the electronic properties of the analyte; (ii) the strength/nature of binding/interaction of the analyte and the binding site, and (iii) as well as the electronic coupling between the binding site and the chromophore as mentioned above. Here we report the synthesis of a chemical probe for anion detection with a binding site constructed from two sulfonamide groups providing H-bonding motif through two N–H bonds, which are suitable to bind to anions. The binding site is mounted on a benzodithiophene fluorophore with two tert-butylphenyl group substituents; the phenyl groups extend the conjugated system of benzodithiophene (shifting its emission to lower energy) and facilitate the solvation of the probe in organic solvents. The binding of an anion to the binding site (through H-bonding with the N–H bonds) increases the electronic density on the N-atoms that are conjugated with the fluorophore. This increase in electron density is distributed over the fluorophore system leading to a blue shift in its emission peak; this shift is correlated with the concentration of the anion in solution.
The synthesis of the probe started by the Suzuki coupling of 2,7-diiodobenzo[2,1-b:3,4-b′]dithiophene-4,5-dione13 (2) with 4-tert-butylphenylboronic acid (3) to afford diketone 4 in 85% yield. Diketone 4 was condensed with N,N′-(4,5-diamino-1,2-phenylene)bis(4-methylbenzenesulfonamide)36 (5) to furnish optical probe 1 in 51% yield (Scheme 1).
Results and discussion
The anion detection properties of optical probe 1 were studied using UV-vis absorbance spectroscopy, fluorescence and 1H NMR titrations with anion solutions in chloroform. The change in the spectroscopic profiles of the optical probe were then monitored and correlated with the anion concentration. The optical properties of 1 in chloroform are shown in Fig. 1; it shows characteristic absorbance peaks at 327 nm, 414 nm, and 498 nm, and an emission peak at 620 nm upon excitation at 420 nm.
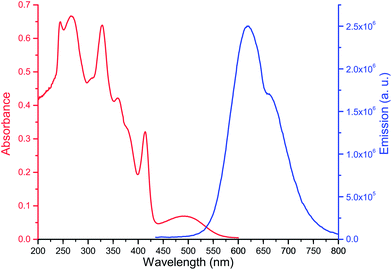 |
| | Fig. 1 Absorbance and emission spectra of 1 (0.01 mM in CHCl3). | |
Effect of anion on absorbance spectra
The addition of anion solutions (1.0 mM in CHCl3) to 1 (0.01 mM in CHCl3) leads to an increase in absorbance at 450 nm and 300 nm concomitant with a decrease at 410 nm and combined with three isosbestic points at 420 nm, 388 nm, and 365 nm. Fig. 2 illustrates the changes observed in the absorption spectra of 1 upon titrating it with a solution of tetrabutylammonium acetate (TBAOAc) solution. This change in spectrum indicates strong binding interaction between the optical probe and the basic anion suggesting strong hydrogen bonding. The relative change of absorbance of 1 at 450 nm upon the increase in the anion concentrations is shown in Fig. 2. Similar spectral changes were observed for the interaction of 1 with benzoate, cyanide and dihydrogen phosphate anions, but insignificant changes were observed for fluoride, chloride, bromide, iodide, and nitrate anions (see ESI, Fig. SI-1†).
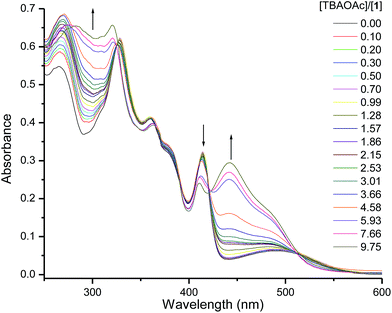 |
| | Fig. 2 UV-vis absorbance spectra of 1 (0.01 mM in CHCl3) upon titrating with TBAOAc solution (1.0 mM). | |
Anion effects on fluorescence
The binding of 1 to anions resulted in major changes in its emission spectra. Titrating optical probe 1 with acetate, benzoate, cyanide, or dihydrogen phosphate anions led to enhancement of the optical probe's emission concurrent with a blue shift of the maximum emission peak from 620 to ∼560 nm. This is an indication of an increase in the electron density on the fluorophore and high-energy charged-transfer emission state of the optical probe–anion complex. Fig. 3 shows the changes in the emission of 1 upon titrating with the cyanide anion, and similar spectra were observed for benzoate, and dihydrogen phosphate. The addition of other anions, such as fluoride, chloride, bromide (see ESI, Fig. SI-2†), iodide, and nitrate had less significant effect on the emission of 1.
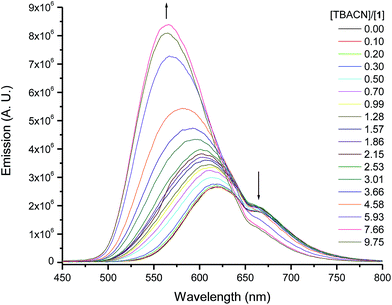 |
| | Fig. 3 Emission spectra of 1 (0.01 mM in CHCl3) upon titrating with TBA cyanide solution (1.0 mM). | |
The relative change in the emission intensity of 1 as a function of the mole ratio of the optical probe to the anion (Fig. 4) is in agreement with the observed trend of the absorbance spectra. The basic anions (acetate, benzoate, cyanide and dihydrogen phosphate) showed the largest change, reaching about 20 times increase in emission at 560 upon addition of four molar equivalents of dihydrogen phosphate anion. It is important to note, however, that the observed trends are unexpected (considering the binding constants, see below) as saturation of signal is not achieved until more than four molar equivalents of the anion have been added. The halides and the nitrate anions showed less significant effect on the emission intensity of 1.
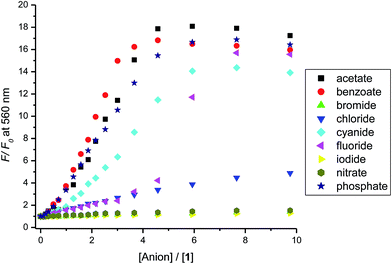 |
| | Fig. 4 Relative change in emission intensity of 1 at 560 nm upon titrating with different anions. | |
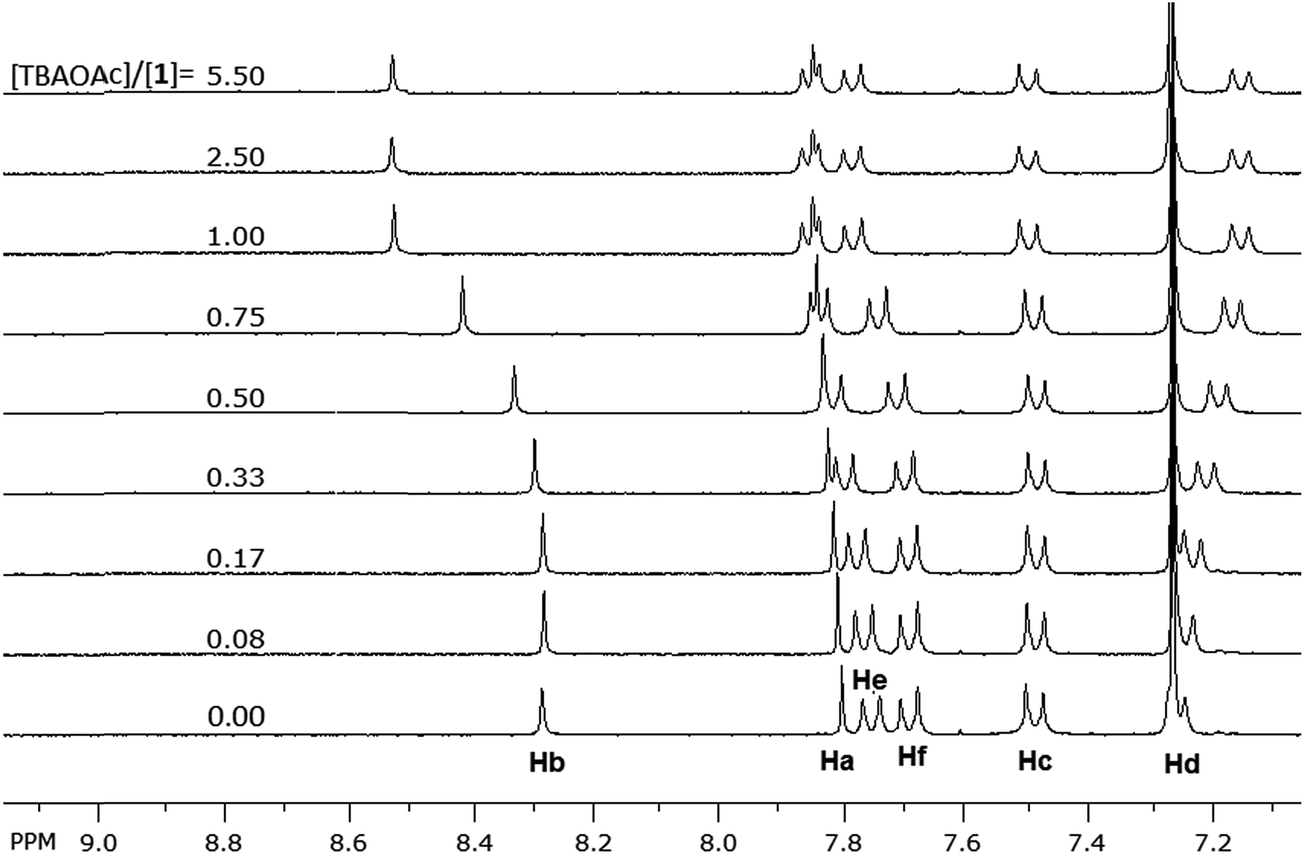
Binding investigation using 1H NMR
1H NMR titrations were conducted on 1 (2.0 mM in CDCl3) with solutions of the anions (20 mM in CDCl3); the signals of N–H protons of the sulfonamide groups, which are associated with the binding process, disappeared from the proton NMR spectrum due to chemical exchange with the residual water in the solvent, thus the signals for the neighboring aromatic protons were monitored. Fig. 5 shows partial NMR spectra for the aromatic protons of 1 at different concentrations of acetate anion; the signals of Ha, Hb, He and Hf (Fig. 6) shifted downfield by different degrees while Hd shifted upfield. The minor downfield shift of Ha is unexpected; previously we observed significant upfield shift for Ha protons in similar compounds due to the increase in the electronic density on the neighboring sulfonamide groups caused by H-bonding of the N–H protons to the anions.29,30 In this case, the downfield shift is attributed to the strong delocalization of electron density on the extended aromatic systems and the electron-withdrawing effect of the sulfur atoms on rings B (Fig. 6) which is induced by the electrostatic interaction of the cation (TBA) with the sulfur atoms. This cation-induced electron-withdrawing effect of sulfur atoms on ring B is supported by the observation of a strong downfield shift of Hb, which is a more significant shift than what we observed previously with optical probes with similar structures,29 and the changes in the chemical shifts of the protons (Hg) on TBA cations (vide infra). The increase of electron density on the N-atom of ring D also contributes to the downfield shift of Hb due to a through-space effect of the electron density on N-atom (forming H-bond-like interaction). The minor downfield shift of Hc and the upfield shift of Hd on F-rings are consistent29 with the increase of electron density on the N-atoms of the sulfonamide groups (which results in a decrease of the electron-withdrawing strength of the sulfonamide group and an increase of the electron density of the oxygen atoms and of the F-rings).
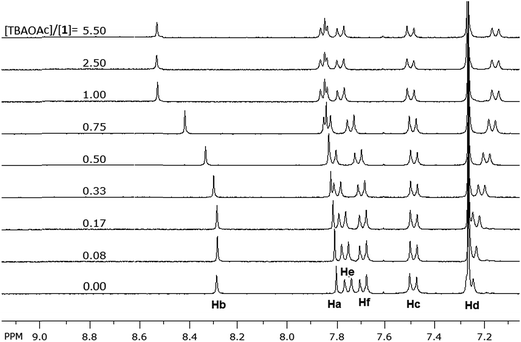 |
| | Fig. 5 Aromatic region of 1H NMR spectra of 1 (2.0 mM) showing the change in the chemical shift of the aromatic protons upon titrating with TBAOAc solution (20 mM) in CDCl3. | |
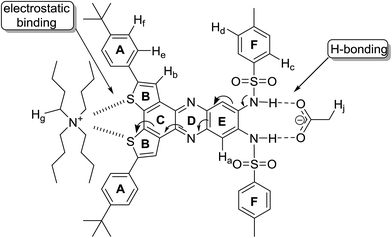 |
| | Fig. 6 Structural representation of the interactions/binding of 1 to the ions of TBAOAc. | |
Both signals of He and Hf shifted downfield upon titrating 1 with the acetate anion; such shifts are consistent with a decrease in the electron density on ring A. This is attributed to the electrostatic interaction of the neighboring sulfur atoms on ring B with TBA cation. This interaction between the sulfur and TBA is suggested by the change, upon titration, in the chemical shifts of the signals corresponding to the protons of TBA cation (similar shifts were also observed for TEA cation, see Fig. SI-3†). Fig. 7 depicts the aliphatic region of the NMR spectra of 1; as a solution of TBAOAc was added into the solution of 1, the signals corresponding to protons of TBA started to appear and increase in intensity. In particular, the signals for Hg protons (Fig. 6), the closest protons to the positively charged nitrogen atom of the TBA, and Hj protons appeared at 3.1 ppm and 2.1 pm respectively. These signals showed insignificant change in the chemical shift until equimolar concentration of 1 and TBAOAc. As more TBAOAc was added (more than 1 equivalent), the signal for Hg shifted downfield while the signal for Hj shifted upfield; this observation is consistent with a decrease of electron density on Hg and an increase of electron density on Hj. At concentrations with one molar equivalent of TBAOAc or less, the acetate is strongly bound to the sulfonamide groups through H-bonding, and hence the electron density on the fluorophore rings increases. Conjugation through the aromatic system (Fig. 6) makes the sulfur atoms of the rings B more electron rich, inducing a strong electrostatic attraction to the positively charged TBA cation. As the molar concentration of TBAOAc increased above one molar equivalent of 1, there were less unbound molecules of 1 and less interaction of the optical probe with either ion; hence, Hg shifted downfield and Hj shifted upfield. This observation suggests that the electrostatic interaction of TBA cation with the optical probe–acetate assembly is stronger than the interaction with the acetate anion, and it could explain the continuous enhancement of optical probe's emission even after one molar equivalent of the anion was added (Fig. 4). It is important to note that the interaction between TBA cation and 1 is significant only when there is a very strong interaction (binding) between the corresponding anion and the N–H bonds of the sulfonamide; hence, it is observed mainly with acetate, benzoate, cyanide and dihydrogen phosphate anions (vide infra).
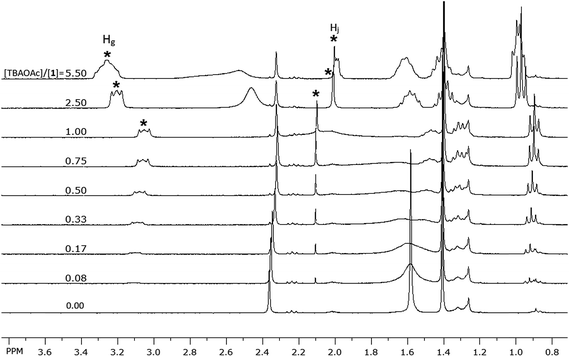 |
| | Fig. 7 Aliphatic region of the 1H NMR spectra of 1 (2.0 mM) showing the change in the chemical shift of the protons of TBA cation upon titrating with TBAOAc solution (20 mM) in CDCl3. | |
In order to estimate the binding constants of the optical probe to the anions, we tried to fit the binding isotherms of Ha signal (Fig. 8) of the investigated ions to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model. The isotherms of the weakly-binding anions (chloride, bromide, iodide and nitrate) fit the model with Ka = 102 to 103 M−1 (Table 1). The binding isotherms of strongly-binding anions (acetate, benzoate, and cyanide), however, showed that the binding constants are too large (Ka > 106 M−1) to be calculated from the NMR titrations.37,38 Therefore, the binding constants of these anions were calculated from the binding isotherms of the absorbance (UV-vis) titrations taking into consideration the binding of the optical probe to both the cation and the anion at the same time (see details in ESI† document). The obtained binding constants (Table 1) showed a strong binding for acetate, benzoate and cyanide anions. This is consistent with the strong enhancement in the emission of 1 observed in the presence of these anions in solution; these anions form hydrogen bonds with the N–H bonds of the sulfonamide groups at the binding site of 1 (each carboxylate anion forms two hydrogen bonds).
:1 binding model. The isotherms of the weakly-binding anions (chloride, bromide, iodide and nitrate) fit the model with Ka = 102 to 103 M−1 (Table 1). The binding isotherms of strongly-binding anions (acetate, benzoate, and cyanide), however, showed that the binding constants are too large (Ka > 106 M−1) to be calculated from the NMR titrations.37,38 Therefore, the binding constants of these anions were calculated from the binding isotherms of the absorbance (UV-vis) titrations taking into consideration the binding of the optical probe to both the cation and the anion at the same time (see details in ESI† document). The obtained binding constants (Table 1) showed a strong binding for acetate, benzoate and cyanide anions. This is consistent with the strong enhancement in the emission of 1 observed in the presence of these anions in solution; these anions form hydrogen bonds with the N–H bonds of the sulfonamide groups at the binding site of 1 (each carboxylate anion forms two hydrogen bonds).
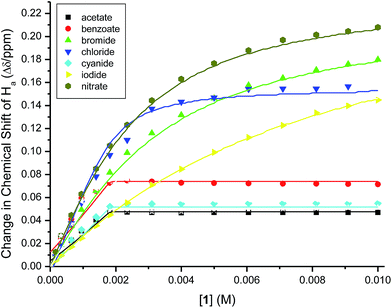 |
| | Fig. 8 The change in the chemical shifts of Ha for 1 (2.0 mM) upon titrating with anion solutions (20 mM) in CDCl3. The solid lines represent the calculated fit of 1:1 binding model. | |
Table 1 Binding constants of optical probe 1 to the corresponding anions
| Anion |
logKa |
| Determined from the changes in the bUV and cNMR spectra upon titration of 1 with the anions. See ESI for more details on Ka calculations. |
| Acetate |
9.252 ± 0.002b |
| Benzoate |
9.525 ± 0.004b |
| Bromide |
2.65 ± 0.03c |
| Chloride |
3.62 ± 0.07c |
| Cyanide |
8.662 ± 0.005b |
| Iodide |
2.17 ± 0.02c |
| Nitrate |
2.84 ± 0.01c |
The binding isotherms (NMR and UV-vis) of the fluoride and dihydrogen phosphate anions did not fit to any of the models discussed above. A closer look at the change in the chemical shift of the signal of Ha protons in 1H NMR upon titrating with fluoride and dihydrogen phosphate anions showed a mixed trend (Fig. 9). The protons shifted downfield upon the addition of the anions up to one molar equivalent; as more anion is added, the signal of Ha protons shifted upfield (more significantly for fluoride anion than for dihydrogen phosphate) until two molar equivalents of the anions have been added. The initial downfield ([anion]/[1] < 1) is due to the strong delocalization of the electron density from ring E to the other rings (vide supra); the subsequent upfield shift (for [anion]/[1] > 1), however, indicates the presence of high electron density on ring E. This suggests a proton transfer (deprotonation) from N–H bonds to the anions; this change in the mechanism of interaction from hydrogen-bonding to proton-transfer was observed previously for basic anions, such as the fluoride anions.39–41 The interaction of fluoride and dihydrogen phosphate anions with binding site also accounts for the observed trend in the shift of the protons on TBA cation (Hg). The change in the signal of Hg protons upon addition of fluoride and dihydrogen phosphate anions (Fig. 9) showed a continuous upfield shift until the addition of about two molar equivalents of each anion, due to the strong interaction of the anions with the optical probe sulfonamide binding site (hydrogen-bonding followed by proton transfer) and strong electrostatic interaction between TBA cation and benzodithiophene rings (B rings) of the optical probe. The subsequent downfield shift in Hg protons is due to the dilution effect upon the increase in concentration of the guest ions. This trend of Hg is also observed for the strong binding anions (acetate, benzoate and cyanide), however, the reverse in the trend from upfield to downfield shift appeared at equimolar mixture of the optical probe and the anion (Fig. 10). The weak-binding anions (chloride, bromide, iodide and nitrate) showed insignificant effect on Hg signal (Fig. 10).
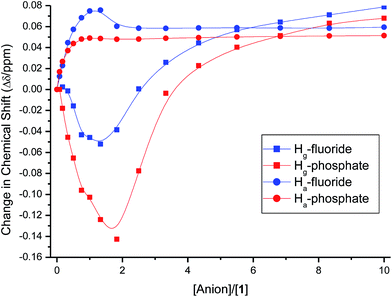 |
| | Fig. 9 The change in the chemical shifts of Ha and Hg upon titrating 1 (2.0 mM) with anion solutions (20 mM) in CDCl3. | |
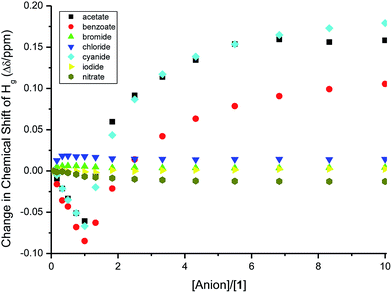 |
| | Fig. 10 The change in the chemical shifts of Hg upon titrating 1 (2.0 mM) with anion solutions (20 mM) in CDCl3. | |
Computational studies
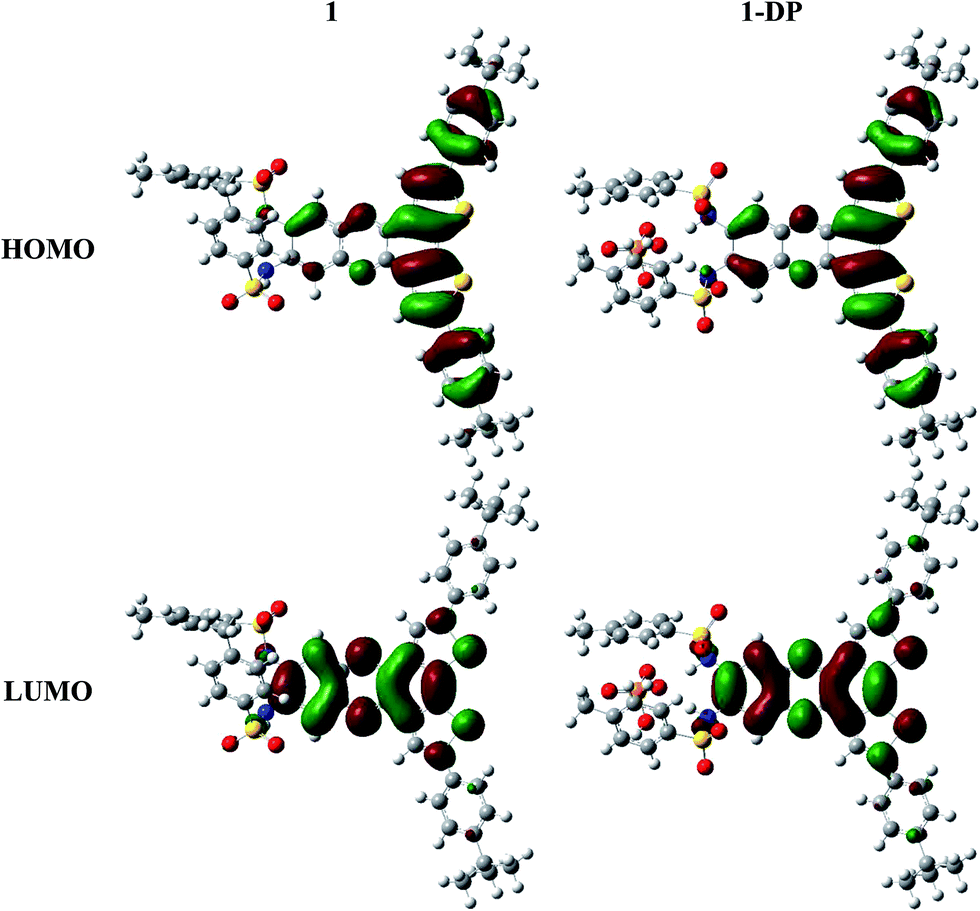
We take for the sake of comparison optical probe 1, first without any anion (1) and then with dihydrogen phosphate (1-DP). Overall the molecular structure is not significantly perturbed by the addition of the anion. Optimization of the geometry confirms that anion binding occurs through hydrogen bonding as the anion resides within expected hydrogen bond distances from the binding site provided by the two sulfonamide groups. The largest geometry change upon anion binding expectedly occurs with the NHTs groups, which rearrange around anion. Simple visual inspection of the Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) reveals very limited modification upon addition of the anion, Fig. 11.
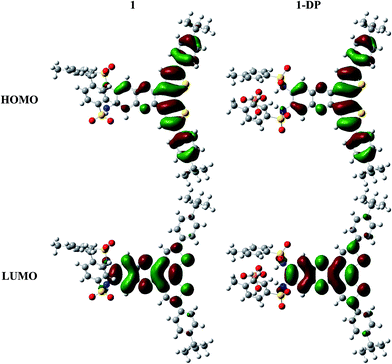 |
| | Fig. 11 DFT-ωB97XD frontier molecular orbitals for optical probe 1 without an anion (left) and in the presence of dihydrogen phosphate (right). | |
Table 2 provides the energies (in eV) for the frontier molecular orbitals. As anticipated, the addition of an anion destabilizes the HOMO and LUMO increasing their energies. This destabilization broadens the HOMO–LUMO gap (ΔE(H–L)) by nearly 0.2 eV. It should be noted that the change in ΔE(H–L) is nearly constant across the series of anions, regardless of the anion's identity, and does not suggest an immediate explanation for the change in the UV-vis spectra.
Table 2 Energies of the DFT-ωB97XD frontier molecular orbitals and HOMO–LUMO energy gap (ΔE(H–L)) for optical probe 1 without an anion (Ø) and for the series of anions which showed strongest binding. All values are reported in eV
| |
Ø |
Cyanide |
Fluoride |
Dihydrogen phosphate |
| LUMO+1 |
0.093 |
0.464 |
0.5246 |
0.429 |
| LUMO |
−1.056 |
−0.518 |
−0.437 |
−0.585 |
| HOMO |
−7.241 |
−6.859 |
−6.802 |
−6.894 |
| HOMO−1 |
−8.057 |
−7.334 |
−7.292 |
−7.523 |
| ΔE(H–L) |
6.184 |
6.336 |
6.364 |
6.308 |
Time-dependent density functional theory was used to probe how the addition of the anion impacts the excited states of the optical probe (Table 3). The presence of the anion does not significantly impact any of the characteristics of the lowest excited state S1 (i.e. energy, transition dipole moment, and oscillator strength); the S1 transition energy remains nearly constant throughout the series. This is consistent with the fact that the S0 → S1 transition essentially corresponds to a HOMO → LUMO transition and the ΔE(H–L) gap remains constant regardless of the identity of the anion.
Table 3 DFT-ωB97XD excited-state energies (in eV) for the first two singlets (Sn) along with corresponding transition dipole moments (μ) from the ground state (S0 → Sn) (in Debye) and oscillator strength (f)
| |
Ø |
Cyanide |
Fluoride |
Dihydrogen phosphate |
| S1 |
3.044 |
3.143 |
3.163 |
3.144 |
| μ(S0–S1) |
5.851 |
6.225 |
6.279 |
6.336 |
| f |
0.395 |
0.462 |
0.473 |
0.478 |
| S2 |
3.361 |
3.422 |
3.421 |
3.509 |
| μ(S0–S2) |
7.235 |
8.293 |
8.191 |
8.577 |
| f |
0.667 |
0.892 |
0.871 |
0.979 |
In fact, the strongest impact upon anion binding is to be found on the second excited state (S2). In general, for 1 both without and with an anion, S2 possesses a stronger transition dipole moment and larger oscillator strength (nearly double) in comparison with S1. While the addition of the anion does perturb the S2 energy, the largest changes are observed with the transition dipole moments and oscillator strength. In the case of dihydrogen phosphate, which showed the most pronounced changes, the transition dipole moment increases from 7.235 D to 8.577 D resulting in an oscillator strength increase by over 0.3. The S0 → S2 transition is dominated by a HOMO−1 → LUMO electron promotion and corresponds to a local excitation involving orbitals located in the central conjugated core of the optical probe, which are not disturbed by the presence of the anion. Thus, it is the second excited state and not the first excited state that is most impacted by the addition of the anion; the increase in the strength of the second optical transition accounts for the observed changes in the UV-vis spectra.
Conclusion
We have synthesized a new dithieno[3,2-a:2′,3′-c]phenazine-based optical probe (1) and investigated its efficacy as a fluorescent chemical optical probe for anions. Spectroscopic studies (absorbance, emission and NMR titrations) have shown strong interaction of the optical probe with carboxylate, dihydrogen phosphate and cyanide anions with anion-binding-induced electrostatic interaction to the TBA counter ion through the thiophene rings. The relatively linear change of emission intensity with the increase of dihydrogen phosphate and cyanide anions concentration before signal saturation suggests that the optical probe can act as a detection probe for these anions. We showed that anions can be used to alter the absorption spectra and the emission intensity of benzodithiophene-based compounds, which can find the application in the fine-tuning of the electronic properties of these materials in optoelectronic and light-harvesting devices. DFT calculations point to changes in the strength of the S0 → S2 transition as responsible for the observed changes in the UV-vis spectra.
Experimental
Chemicals and solvents were purchased from Acros. Standard grade silica gel (60 Å, 32–63 μm) and silica gel plates (200 μm) were purchased from Sorbent Technologies. Reactions that required anhydrous conditions were carried out under argon in oven-dried glassware. A Bruker spectrometer was used to record the NMR spectra. CDCl3 was the solvent for NMR and chemical shifts relative to TMS at 0.00 ppm are reported in parts per million (ppm) on the δ scale. The MALDI data were acquired at the Bioanalytical Mass Spectrometry Facility at the Georgia Institute of Technology, Atlanta, GA. Elemental analysis were performed at Atlantic Microlab Inc., Norcross, GA. Absorption spectra were measured using a JASCO V-570 UV-VIS-NIR Spectrophotometer, and the fluorescence measurements were done using a Jobin-Yvon-Horiba Fluorolog III spectrofluorometer. The excitation source was a 100 W xenon lamp, and the detector used was R-928 operating at a voltage of 950 V.
2,7-Diiodobenzo[1,2-b:3,4-b′]dithiophene-4,5-dione13 (2) and 1,2-bis(p-methylphenylsulfonamido)-4,5-diaminobenzene36 (5) were synthesized according to literature procedures.
2,7-Bis(4-(tert-butyl)phenyl)benzo[2,1-b:3,4-b′]dithiophene-4,5-dione (4)
A mixture of 2,7-diiodobenzo[1,2-b:3,4-b′]dithiophene-4,5-dione (2) (0.20 g, 0.42 mmol) in 20 mL of anhydrous 1,4-dioxane was purged with argon for 15 min, then 4-tert-butylphenylboronic acid (3) (0.23 g, 1.27 mmol, 3 eq.) and anhydrous potassium carbonate (0.7 g, 5.07 mmol) was added. Argon gas was bubbled through the solution for 30 min, then Pd(PPh3)4 (50 mg, 0.04 mmol) was added, and the reaction mixture was purged with argon for additional 15 min. The reaction was left to stir at 80 °C for 3 days under argon. After 3 days, the reaction mixture was poured into ice, and hydrochloric acid (37% concentration) was added. The organic matter was extracted with dichloromethane (DCM), combined organic phases were dried over anhydrous magnesium sulphate, and the drying agent was removed by filtration. Solvent was removed under reduced pressure, and a black solid was obtained. This solid was purified by column chromatography using (hexanes:DCM (3:1)) as eluent, and the product (4) was obtained as a dark blue solid (0.17 g, 85%). 1H NMR (500 MHz, CDCl3): δ 7.58 (s, 2H), 7.48 (d, J = 8.5 Hz, 4H), 7.40 (d, J = 8.5 Hz, 4H), 1.29 (s, 18H). 13C{1H} NMR (125 MHz, CDCl3): δ 174.65, 152.51, 144.88, 142.38, 135.84, 129.35, 126.22, 125.55, 122.11, 34.80, 31.17. HR-MALDI (m/z): [M]+: calcd for C30H28O2S2, 484.1531; found, 484.1516. Anal. calcd for C30H28O2S2: C, 74.35; H, 5.82; S, 13.23. Found: C, 74.25; H, 5.97; S, 13.06.
N,N′-(2,5-bis(4-(tert-butyl)phenyl)dithieno[3,2-a:2′,3′-c]phenazine-9,10-diyl)bis(4-methylbenzenesulfonamide) (1)
A mixture of ethanol and acetic acid (1:1, 40 mL) was added to a flask containing 1,2-bis(p-methylphenylsulfonamido)-4,5-diaminobenzene (5) (1.00 g, 2.23 mmol (excess)). This mixture was purged with argon for 15 minutes, then 4 (0.16 g, 0.33 mmol) was added, the mixture was purged with argon for additional 15 minutes, and then the reaction was left to stir at 105 °C for 3 days. After three days, the reaction was cooled, vacuum filtered, and an orange solid was obtained. The crude material was recrystallized from toluene, and 1 was obtained as orange reddish crystals (0.15 g, 51%). 1H NMR (CDCl3, 500 MHz): δ 8.11 (s, 2H), 7.70 (d, J = 8.0 Hz, 4H), 7.67 (s, 2H), 7.59 (d, J = 8.0 Hz, 4H), 7.49 (bs, 2H), 7.41 (d, J = 8.5 Hz, 4H), 7.19 (d, J = 8.5 Hz, 4H), 2.28 (s, 6H), 1.33 (s, 18H). 13C{1H} NMR (CDCl3, 125 MHz): δ 151.65, 144.63, 143.38, 139.57, 139.47, 134.92, 134.82, 132.12, 130.79, 129.89, 127.77, 126.03, 125.93, 123.06, 119.40, 34.77, 31.28, 21.62. HR-MALDI (m/z): [M]+: calcd for C50H46N4O4S4, 894.2395; found, 894.2408. Anal. calcd for C50H46N4O4S4: C, 67.09; H, 5.18; N, 6.26; S, 14.33. Found: C, 67.38; H, 5.40; N, 6.17; S, 14.03.
Spectroscopic titration. A solution of the optical probe (1) (10 μM, 2 mL) in CHCl3 placed into a 1 × 1 cm quartz cuvette was titrated with a solution of the anion (1.0 mM in CHCl3) that contained the optical probe (10 μM). Aliquot amounts of the anion solution were added to the cuvette via a syringe until a total of ten or more equivalents of the anion had been added (the number of additions was around 20 with an increase in the amount of anion solution added). The UV-vis spectrum and emission spectrum (λex = 420 nm) were scanned after each addition.
1H NMR titration. A solution of optical probe (1) (2 mM, 600 μL) in CDCl3 placed in an NMR tube was titrated with a solution of the anion (20 mM). Aliquot amounts of the anion solution were added to the NMR tube via a syringe until a total of ten equivalents of the anion were added (the number of additions was around 17 with an increase in the amount of anion solution added). 1H NMR spectrum was recorded after each addition, and the chemical shifts of the protons were recorded. The collected data were analyzed using a non-linear least square regression program to fit the data to a theoretical model of a 1:1 binding.
Computational methodology
Ground-state geometry optimizations of optical probe 1 without and with anions were performed with Density functional theory (DFT) using the ωB97XD functional42 and cc-pVDZ basis set.43 The absence of imaginary frequencies was used to confirm that the geometries corresponded to minima on the ground-state potential energy surface. Time-dependent DFT was used to obtain the vertical excited-state energies for the lowest 5 singlet and 5 triplet states at the optimized ground-state (S0) geometry. The calculations were conducted in the presence of an implicit solvent using the Polarizable Continuum Model, using the dielectric constant of chloroform for direct comparison to experiment. All calculations were performed using the Gaussian09 code.44
Acknowledgements
This work was supported by the Lebanese National Council for Scientific Research (CNRS). The authors are grateful for this support. B. R. K. thanks the Arab Fund Scholarship Program for financial support. M. H. A. thanks the financial support (FRG10-02 and FRG12-3-18) of the American University of Sharjah, UAE. S. R. M. thanks the Georgia Power Foundation and Solvay SA for support. J. L. B. acknowledges support from the King Abdullah University of Science & Technology.
References
- F. De Riccardis, I. Izzo, D. Montesarchio and P. Tecilla, Acc. Chem. Res., 2013, 46, 2781–2790 CrossRef CAS PubMed.
- A. Vargas Jentzsch, A. Hennig, J. Mareda and S. Matile, Acc. Chem. Res., 2013, 46, 2791–2800 CrossRef CAS PubMed.
- T. Schraderr and A. D. Hamilton, Functional Synthetic Receptors, Wiley-VCH, Weinheim, Germany, 2005 Search PubMed.
- W. J. Marshall and S. K. Bangert, Clinical Chemistry, Elsevier, Edinburgh, 5th edn, 2004 Search PubMed.
- R. F. Schmidt and G. Thews, Human Physiology, Springer-Verlag, Berlin, 2nd edn, 1989 Search PubMed.
- J. L. Sessler, P. A. Gale and W. S. Cho, Anion Receptor Chemistry, Royal Society of Chemistry, Cambridge, UK, 2006 Search PubMed.
- J. Hladilkova, Z. Prokop, R. Chaloupkova, J. Damborsky and P. Jungwirth, J. Phys. Chem. B, 2013, 117, 14329–14335 CrossRef CAS PubMed.
- M. L. Matthews, W.-C. Chang, A. P. Layne, L. A. Miles, C. Krebs and J. M. Bollinger Jr, Nat. Chem. Biol., 2014, 10, 209–215 CrossRef CAS PubMed.
- D. Vullo, E. V. Kupriyanova, A. Scozzafava, C. Capasso and C. T. Supuran, Bioorg. Med. Chem., 2014, 22, 1667–1671 CrossRef CAS PubMed.
- Z. Xu, X. Chen, H. N. Kim and J. Yoon, Chem. Soc. Rev., 2010, 39, 127–137 RSC.
- A. Mitra, V. K. Hinge, A. Mittal, S. Bhakta, P. Guionneau and C. P. Rao, Chem.–Eur. J., 2011, 17, 8044–8047 CrossRef CAS PubMed.
- Y. Nishide, H. Osuga, M. Saito, T. Aiba, Y. Inagaki, Y. Doge and K. Tanaka, J. Org. Chem., 2007, 72, 9141–9151 CrossRef CAS PubMed.
- Y. A. Getmanenko, M. Fonari, C. Risko, B. Sandhu, E. Galan, L. Zhu, P. Tongwa, D. K. Hwang, S. Singh, H. Wang, S. P. Tiwari, Y.-L. Loo, J.-L. Brédas, B. Kippelen, T. Timofeeva and S. R. Marder, J. Mater. Chem. C, 2013, 1, 1467–1481 RSC.
- C. Taliani, R. Danieli, R. Zamboni, G. Giro and F. Sannicolo, Springer Ser. Solid-State Sci., 1987, 76, 326–329 CAS.
- I. Akimoto, K.-I. Kan'no, H. Osuga and K. Tanaka, J. Lumin., 2005, 112, 341–344 CrossRef CAS PubMed.
- N. Hundt, K. Palaniappan, J. Servello, D. K. Dei, M. C. Stefan and M. C. Biewer, Org. Lett., 2009, 11, 4422–4425 CrossRef CAS PubMed.
- Y. A. Getmanenko, L. E. Polander, D. K. Hwang, S. P. Tiwari, E. Galan, B. M. Seifried, B. Sandhu, S. Barlow, T. Timofeeva, B. Kippelen and S. R. Marder, J. Org. Semicond., 2013, 1, 7–15 CrossRef CAS PubMed.
- Y. Chen, Y. Yan, Z. Du, X. Bao, Q. Liu, V. A. L. Roy, M. Sun, R. Yang and C. S. Lee, J. Mater. Chem. C, 2014, 2, 3921–3927 RSC.
- Y. Didane, G. H. Mehl, A. Kumagai, N. Yoshimoto, C. Videlot-Ackermann and H. Brisset, J. Am. Chem. Soc., 2008, 130, 17681–17683 CrossRef CAS PubMed.
- D. Liu, M. Xiao, Z. Du, Y. Yan, L. Han, V. A. L. Roy, M. Sun, W. Zhu, C. S. Lee and R. Yang, J. Mater. Chem. C, 2014, 2, 7523–7530 RSC.
- H. D. Magurudeniya, R. S. Kularatne, E. A. Rainbolt, M. P. Bhatt, J. W. Murphy, E. E. Sheina, B. E. Gnade, M. C. Biewer and M. C. Stefan, J. Mater. Chem. A, 2014, 2, 8773–8781 CAS.
- J. Min, Z.-G. Zhang, S. Zhang and Y. Li, Chem. Mater., 2012, 24, 3247–3254 CrossRef CAS.
- R. Rieger, D. Beckmann, A. Mavrinskiy, M. Kastler and K. Müllen, Chem. Mater., 2010, 22, 5314–5318 CrossRef CAS.
- S. Wang, S. Ren, Y. Xiong, M. Wang, X. Gao and H. Li, ACS Appl. Mater. Interfaces, 2013, 5, 663–671 CAS.
- J. Yin, Y. Zhou, T. Lei and J. Pei, Angew. Chem., Int. Ed., 2011, 50, 6320–6323 CrossRef CAS PubMed.
- H.-J. Yun, M. C. Hwang, S. M. Park, R. Kim, D. S. Chung, Y.-H. Kim and S.-K. Kwon, ACS Appl. Mater. Interfaces, 2013, 5, 6045–6053 CAS.
- Y. Liang and L. Yu, Acc. Chem. Res., 2010, 43, 1227–1236 CrossRef CAS PubMed.
- L. Ye, S. Zhang, L. Huo, M. Zhang and J. Hou, Acc. Chem. Res., 2014, 47, 1595–1603 CrossRef CAS PubMed.
- F. S. Raad, A. O. El-Ballouli, R. M. Moustafa, M. H. Al-Sayah and B. R. Kaafarani, Tetrahedron, 2010, 66, 2944–2952 CrossRef CAS PubMed.
- A. O. El-Ballouli, Y. Zhang, S. Barlow, S. R. Marder, M. H. Al-Sayah and B. R. Kaafarani, Tetrahedron Lett., 2012, 53, 661–665 CrossRef CAS PubMed.
- N. V. Ghule, S. V. Bhosale and S. V. Bhosale, RSC Adv., 2014, 4, 27112–27115 RSC.
- H. Zhang, P. Wang, Y. Yang and H. Sun, Chem. Commun., 2012, 48, 10672–10674 RSC.
- T. Ema, K. Okuda, S. Watanabe, T. Yamasaki, T. Minami, N. A. Esipenko and P. Anzenbacher Jr, Org. Lett., 2014, 16, 1302–1305 CrossRef CAS PubMed.
- S. V. Bhosale, S. V. Bhosale, M. B. Kalyankar and S. J. Langford, Org. Lett., 2009, 11, 5418–5421 CrossRef CAS PubMed.
- T.-P. Lin, C.-Y. Chen, Y.-S. Wen and S.-S. Sun, Inorg. Chem., 2007, 46, 9201–9212 CrossRef CAS PubMed.
- A. Kleineweischede and J. Mattay, Eur. J. Org. Chem., 2006, 947–957, DOI:10.1002/ejoc.200500548.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- L. Fielding, Tetrahedron, 2000, 56, 6151–6170 CrossRef CAS.
- V. Amendola, D. Esteban-Gomez, L. Fabbrizzi and M. Licchelli, Acc. Chem. Res., 2006, 39, 343–353 CrossRef CAS PubMed.
- C.-Y. Wu, M.-S. Chen, C.-A. Lin, S.-C. Lin and S.-S. Sun, Chem.–Eur. J., 2006, 12, 2263–2269 CrossRef CAS PubMed.
- L. S. Evans, P. A. Gale, M. E. Light and R. Quesada, New J. Chem., 2006, 30, 1019–1025 RSC.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, Revision B.01 edn, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Absorption and emission spectra upon titrating with tetrabutylammonium anion solution, change in the chemical shift of Hg using TBABr vs. TEABr, calculations of binding constants, and the 1H and 13C spectra of 1 and 4 are provided. See DOI: 10.1039/c5ra01416f |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.