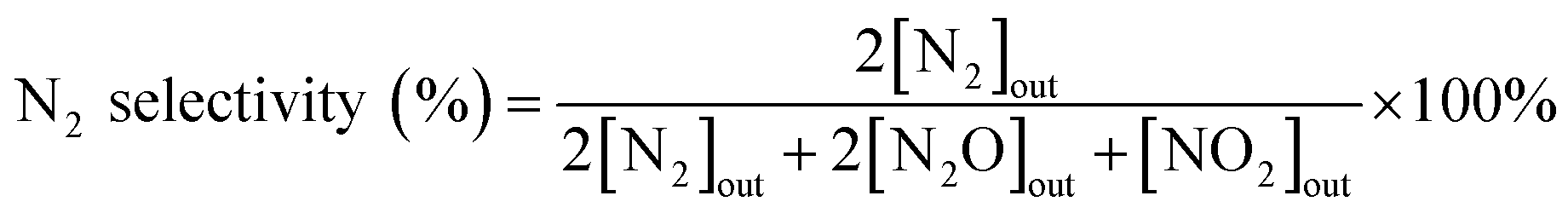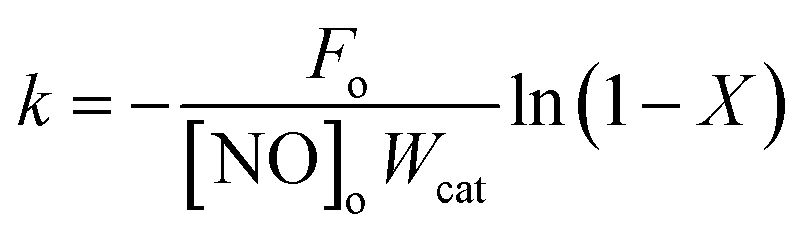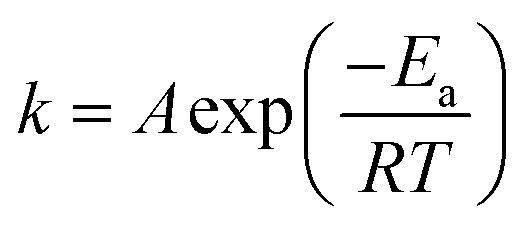DOI:
10.1039/C5RA05978J
(Paper)
RSC Adv., 2015,
5, 45172-45183
Direct synthesis of V–W–Ti nanoparticle catalysts for selective catalytic reduction of NO with NH3
Received
3rd April 2015
, Accepted 15th May 2015
First published on 15th May 2015
Abstract
A series of V–W–Ti nanoparticle catalysts with variable V doping amounts were directly synthesized by the sol–gel method, and their catalytic performances were tested for the selective catalytic reduction of NO with ammonia. The catalysts were characterized by means of XRD, Raman, BET, TEM, SEM, NH3-TPD, H2-TPR and XPS. SCR kinetic studies were conducted to understand the mechanistic features of V–W–Ti catalysts. It was found that the V0.02W0.04Ti catalyst exhibited the highest NO conversion and the lowest apparent activation energy. The characterization results showed that V was incorporated into the TiO2 framework and the redox cycle of V4+ + Ti4+ → V5+ + Ti3+ existed over the V–W–Ti catalysts. A high concentration of reducible and distorted V species could account for the excellent NH3-SCR catalytic performance of the V0.02W0.04Ti catalyst. In situ FT-IR spectroscopy was performed to investigate the mechanism of the NH3-SCR reaction over the V0.02W0.04Ti catalyst. Experimental results showed that both Lewis and Brønsted acid sites over the V0.02W0.04Ti catalyst were involved in the NH3-SCR reaction. The adsorption of nitrate species was significantly limited and the adsorbed NO2 gaseous molecules were easily formed over the V0.02W0.04Ti catalyst, which resulted in the high catalytic activity at low temperature.
1. Introduction
Over the past decades, nitrogen oxides (NOx, including NO, NO2 and N2O) from stationary and mobile sources have remained as major air pollutants. NOx emissions have direct negative effects on the human respiratory system, due to the formation of acid rain, photochemical smog and ozone depletion.1,2 In recent years, the NOx emission standard is becoming more and more stringent worldwide. Therefore, it is necessary to develop advanced technology and seek better catalysts to control NOx emissions.
The selective catalytic reduction (SCR) of NO by NH3 in the presence of excess oxygen is one of the best world technologies to remove NOx from stationary sources and diesel engines. The commonly used commercial catalysts for NH3-SCR are based on V2O5–WO3/TiO2-based catalysts owing to its highly catalytic activity and sulfur tolerance under actual flue-gas conditions. However, these kinds of catalysts are only efficient in a narrow temperature window (300–400 °C).3–5 Generally, titanium dioxide (TiO2) supporters and vanadium(V) are used as active catalytic materials. With the increasing of vanadium loading on titania surface, vanadium species are transformed from tetrahedral VO4 (ref. 6 and 7) to partly distorted dimeric or oligomeric species,8,9 then to isolated polymer.10 Finally, crystalline V2O5 is present when vanadium loading amount is beyond the theoretical monolayer coverage.
Most V-based catalysts were prepared by solution impregnation method,11–13 and the dispersion would decline at high vanadium loading, and amorphous V species would transform to crystalline one. Thus, the limit of the dispersion would lead to the decrease of NH3-SCR activity. In this work, sol–gel method is used to introduce vanadium active sites into the framework of TiO2 supports and improve its catalytic activity. The catalysts, which exhibited highly dispersed active species and in which the active metals were existed in a tetrahedral coordination structure, have higher activity than those prepared by an impregnation or precipitation method. Moreover, TiO2 support is modified by doping another component. Many reports are focused on the effects of W in V-based catalysts. Shan et al.14 and Peng et al.15 reported that the addition of WO3 can provide much more Brønsted acidic sites compared with other metal oxides, which is thought to be the reason why the activity are higher than that of other metal oxides. Cristallo et al.16 revealed that the suitable amount of WO3 can stabilize the anatase phase of TiO2 and preserve the surface characteristic of the catalysts. Recently, Kompio et al.17 pointed a new view that the promotional effect of tungsten may originate from the neighboring vanadium oxide species or from the disruption of surface vanadium oxide aggregating, which is beneficial to the SCR performance.
In present work, we synthesized a series of V–W–Ti nanoparticle catalysts with variable V doping amounts by the sol–gel method. Their catalytic performances were tested for selective catalytic reduction of NO with ammonia and the activation energy was calculated to describe the activity of catalysts. NH3-SCR mechanism of these catalysts was also investigated systematically.
2. Experimental
2.1. Catalyst preparation
V–W–Ti nanoparticle catalysts were prepared by the sol–gel method. In a typical synthesis, 34 mL of deionized water, 34 mL of anhydrous ethanol and 6.8 mL of nitric acid were mixed in the break (denoted as solution A), and then a certain amount of ammonium metavanadate and ammonium tungstate were dissolved in solution A (denoted as solution B). The solution of butyl titanate (34 mL) and anhydrous ethanol (136 mL) were added dropwisely to solution B under vigorous stirring to carry out hydrolysis. After continuously strirring for 3 h, the yellowish transparent sol was yielded. Subsequently, the sol was dried at 80 °C for 24 h to form xerogel. After being crushed and sieved to 40–60 mesh, the xerogel was calcinated at 500 °C for 5 h in air. For the sake of the brevity, the catalysts were denoted as VxW0.04Ti. Where, x and 0.04 represented the molar ratio of V/Ti and W/Ti, respectively, and x = 0, 0.004, 0.012, 0.02, 0.028, 0.036.
2.2. Catalyst characterization
The crystal structures of the fresh catalysts were determined with a powder X-ray diffractometer, using Cu Ka (λ = 0.1542 nm) radiation combined with a nickel filter operating at 40 kV and 10 Ma (Shimadzu, Japan). The diffractometer data were recorded for 2θ values from 10° to 80° at a scanning rate of 4° min−1. The patterns were compared with ICDD files for phase identification.
The Brunauer–Emmett–Teller (BET) surface areas were measured by N2 adsorption and desorption (Quantachrome Instruments, USA). The samples were degassed at 200 °C for 12 h.
The TEM images were carried out using a JEILJEM 2100 electron microscope equipped with a field emission source at an accelerating voltage of 200 kV. Drops of the suspension were applied, and after drying, the fine particles were well dispersed on a copper grid coated with carbon.
The surface morphology of the catalyst was observed by field emission scanning electron microscopy (FESEM) on a Quanta 200F instruments using accelerating voltages of 5 kV, in combination with an EDAX genesis 4000 energy-dispersive X-ray spectrometer (EDX). Samples for SEM were dusted on an adhesive conductive carbon belt attached to a copper disk and were coated with 10 nm Au prior to measurement.
X-ray photoelectron spectroscopy (XPS) spectra were recorded with a standard AlK source (1486.6 eV) working at 350 W (XSAM800, Kratos Analytical company, UK). The working pressure was less than 2 × 10−7 Pa. The spectrometer was calibrated by assuming the binding energy (BE) of the Au 4f7/2 line to lie at 84.0 eV with respect to the Fermi level. Binding energies of Ti 2p, Ce 3d, O 1s were calibrated using the C 1s peak (BE = 284.8 eV) as standard.
The laser Raman experiments were performed at room temperature (Renishaw, UK). A laser radiating at 532 nm was used as the excitation source, and the laser power of below 0.07 mW was applied at the sample. Before measurements, the samples were well ground and mounted into a spinning holder to avoid thermal damage during the scanning.
Temperature programmed reduction with H2 (H2-TPR) experiments were performed on the Quantachrome Instruments of autosorb iQ. 100 mg sample was pretreated under Ar by calcination at 300 °C for 1 h and subsequently cooled to 30 °C. Afterwards, 10% H2/Ar flow (60 mL min−1) was passed over the catalyst bed while the temperature was ramped from 30 to 800 °C at a heating rate of 10 °C min−1.
Temperature-programmed desorption of ammonia (NH3-TPD) was performed in a conventional flow apparatus. Each sample was pretreated with N2 at 500 °C for 1 h, then saturated with high-purity anhydrous ammonia at 100 °C for 1 h and subsequently flushed at the same temperature for 1 h to remove physisorbed ammonium. Finally, the TPD operation was carried out from 100 to 700 °C at a heating rate of 10 °C min−1. The amount of NH3 desorbed was monitored by a thermal conductivity detector (TCD).
In situ DRIFTS spectra were recorded using a thermo Nicolet IS50 spectrometer, which was equipped with a high temperature environmental cell fitted with ZnSe window and an MCT detector cooled with liquid N2. The catalyst was loaded in the Harrick IR cell and heated to 400 °C under N2 at a total flow rate of 50 cm3 min−1 for 60 min to remove adsorbed impurities. A background spectrum was collected under a flowing N2 atmosphere and was subtracted from the sample spectra. The DRIFTS spectra were recorded by accumulating 32 scans with a resolution of 4 cm−1.
2.3. Catalytic performance test
The activities of various catalysts for the NH3-SCR of NO were carried out in a fixed bed quartz reactor (i.d. 6 mm). The feed gas mixture consisted of 1000 ppm of NO, 1000 ppm of NH3, 3 vol% of O2. A total flow rate of 500 mL min−1 was maintained for all experiments with N2 as the balance gas. The concentration of NO in the inlet and outlet gas was measured by a flue gas analyzer (Model-4000VM, SIGNAL International Ltd., UK). Meanwhile, the concentration of NH3, NO, NO2 and N2O were measured by a FTIR spectrometer (Nicolet IS50). All catalysts were kept on stream at each temperature for 1/2 h. The NO conversion was defined as follow:| |
 | (1) |
where NO(inlet) represents the NO concentration in the inlet (ppm), NO(outlet) represents the NO concentration in the outlet (ppm).
The N2 selectivity in SCR reaction was calculated from eqn (2).
| |
 | (2) |
3. Results and discussion
3.1. NH3-SCR performance
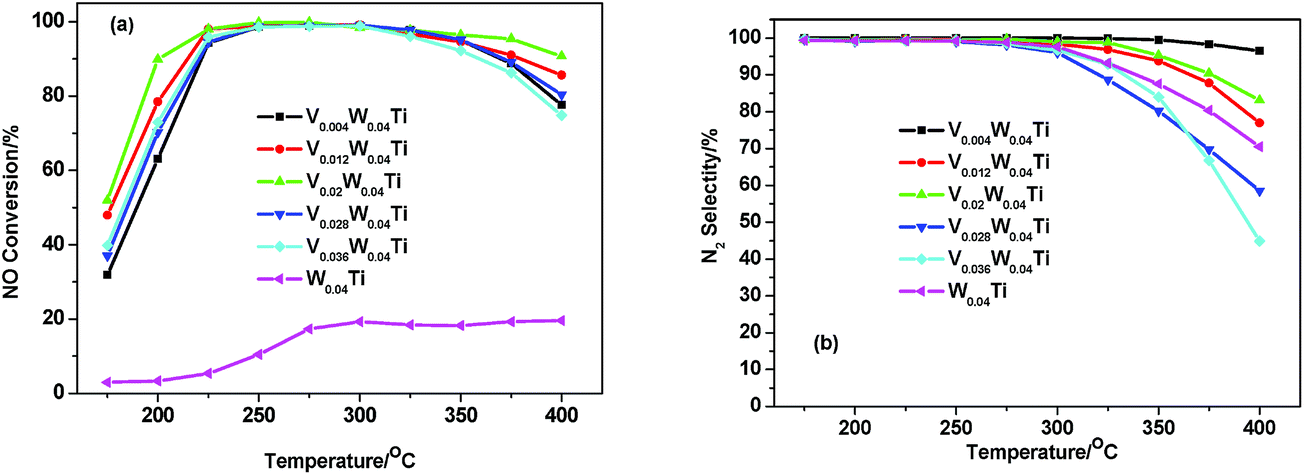
NH3-SCR activities of V–W–Ti catalysts were evaluated as a function of temperature and the results are presented in Fig. 1a. V–W–Ti catalysts showed relatively high activity, while W–Ti catalyst exhibited nearly no activity in the temperature range of 200–400 °C. This result suggests that vanadium oxide species be the main activity phase of V–W–Ti catalysts. The V doping at an appropriate amount enhanced NO conversion and widened the active temperature range. With the increasing of V doping molar amount from 0 to 0.02, the SCR activity showed a monotonic increase. Further increasement of the vanadium doping amount to 0.036 led to the decrease of the activity. For V0.02W0.04Ti catalyst, NO conversion was obtained at 90% in a wide temperature range (200–400 °C) under a GHSV of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. N2 selectivities of all samples were above 95% irrespective of the catalytic activity below 300 °C (Fig. 1b). The N2 selectivity showed a decrease above 300 °C with the increasing of vanadium content, which was mainly due to the occurrence of unselective catalytic oxidation of NH3. V0.02W0.04Ti catalyst is an optimal NH3-SCR catalyst at low temperature.
000 h−1. N2 selectivities of all samples were above 95% irrespective of the catalytic activity below 300 °C (Fig. 1b). The N2 selectivity showed a decrease above 300 °C with the increasing of vanadium content, which was mainly due to the occurrence of unselective catalytic oxidation of NH3. V0.02W0.04Ti catalyst is an optimal NH3-SCR catalyst at low temperature.
 |
| | Fig. 1 NO conversion (a) and N2 selectivity (b) for V–W–Ti catalysts as a function of reaction temperatures. Reaction conditions: [NO] = [NH3] = 1000 ppm, [O2] = 3%, balance N2, total gas flow rate 500 mL min−1, and GHSV = 50000 h−1. | |
3.2. Kinetics analysis of V–W–Ti catalysts on NH3-SCR reactivity
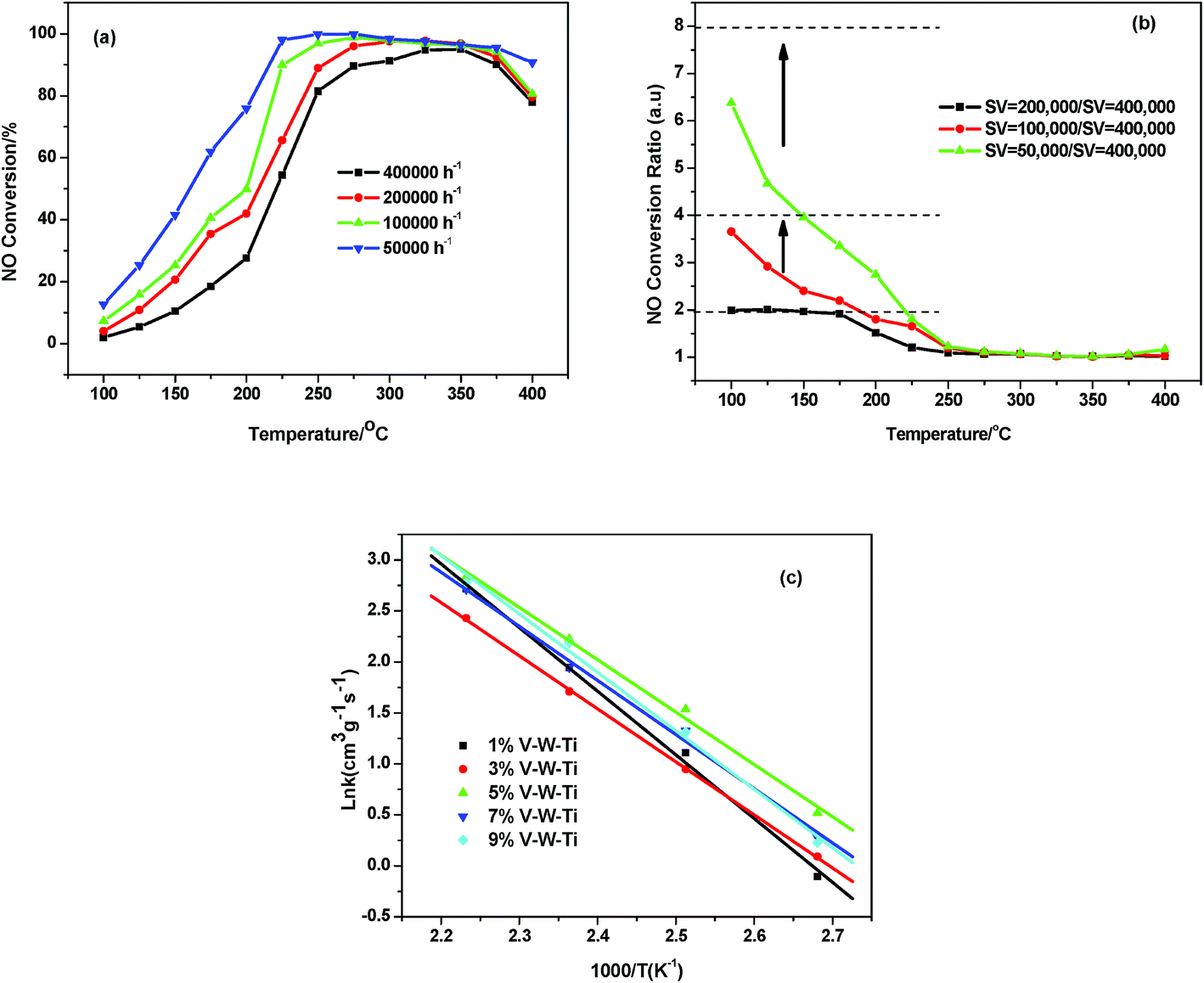
To understand the intrinsic effect of vanadium doping on the activity of V–W–Ti catalysts, the catalytic activity data are quantitatively analyzed using the macro-kinetic approach. To avoid the diffusive effect during the reaction, the influence of different space velocity on the NH3-SCR activity of V0.02W0.04Ti catalyst has been studied. As shown in Fig. 2a, the NO conversion decreased at whole temperature window as the GHSV increased from 50000 to 400000 h−1, which is due to the more contact time for reactant gas to react on the catalysts. According to the previous study,18,19 it was reasonable to assume this reactor as a differential reactor when the NO conversion is below 25% and in the absence of inter-particle mass transfer limitations, NO conversion is expected to increase n-fold as the space velocity decreases n-fold. Based on this criterion, NOx conversion ratio (a ratio of NOx conversion at different space velocity to that at GHSV = 400000 h−1) is shown in Fig. 2b. In this investigation, for catalysts, interparticle mass-transfer limitation can be ignored under GHSV ≥ 400000 h−1 and temperature ≤175 °C conditions.
 |
| | Fig. 2 (a) NO conversion versus reaction temperature for NH3-SCR over V0.02W0.04Ti sample at various space velocities from 50000 to 400000 h−1; (b) NO conversion ratios with respect to that at GHSV = 400000 h−1 as a function of reaction temperature, using data from (a); (c) Arrhenius plots of intrinsic reaction rate constants of the catalysts. | |
The NH3-SCR on the V based catalysts are generally considered to be a first-order reaction with respect to NO.20 Assuming the diffusion-limitation free, the effective first-order rate constant is related to NO conversion (X) by
| |
 | (3) |
| |
 | (4) |
where
k is the reaction constant (cm
3 g
−1 s
−1);
Fo is the molar NO feed rate (mol s
−1); [NO]
o is the molar NO concentration at the inlet (mol cm
−3);
Wcat is the catalyst weight (g);
X is the conversion of NO (%);
A is the pre-exponential factor;
Ea is the apparent activation energy (kJ mol
−1);
R is the gas constant (8.3145 J mol
−1 K
−1); and
T is the temperature (K).
Fig. 2c shows the Arrhenius plots based on the reaction between 100 and 175 °C. The A and Ea are determined from these plots based on eqn (4) and given in Table 1. Ea values of all samples are above 40 kJ mol−1,21 which indicated that the NH3-SCR process at low temperature was controlled by chemical reaction control. Ea values gradually decrease with the increasing of vanadium molar contents from 0.004 to 0.02. Further increasing vanadium content led to Ea values increasement.
Table 1 Kinetic parameter (A and Ea) for NH3-SCR reaction over the catalysts
| Sample |
V0.004W0.04Ti |
V0.012W0.04Ti |
V0.02W0.04Ti |
V0.028W0.04Ti |
V0.036W0.04Ti |
| Ea (kJ mol−1) |
51.5 |
43.2 |
42.4 |
44.1 |
47.4 |
| A |
1.8 × 107 |
1.2 × 106 |
1.6 × 106 |
2.2 × 106 |
6 × 106 |
3.3. N2 physisorption results
Table 2 displays the textural and structural properties of V–W–Ti catalysts. The surface area and pore volume showed a monotonic decrease with the increasing of V doping amount and V0.036W0.04Ti catalyst was of the lowest surface area. All V–W–Ti catalysts exhibited pore sizes in the range of 10–30 nm, indicating that this series of catalysts prepared by the sol–gel method were present as mesoporous materials. Although the BET surface area of V0.02W0.04Ti catalyst was lower than that of V0.004W0.04Ti catalyst, it exhibited higher NH3-SCR activity. This result indicates that the BET surface area did not play a key role in the SCR reaction.
Table 2 The textural and structural properties of V–W–Ti catalysts
| Catalysts |
SBETa (m2 g−1) |
Vmicb (cm3 g−1) |
Average pore diameter (nm) |
| Calculated by BET method. Calculated by t-plot method. |
| W0.04Ti |
60.3 |
1.477 |
17.25 |
| V0.004W0.04Ti |
57.2 |
1.366 |
16.80 |
| V0.012W0.04Ti |
49.5 |
1.016 |
18.14 |
| V0.02W0.04Ti |
45.4 |
0.866 |
12.72 |
| V0.028W0.04Ti |
39.5 |
0.617 |
21.79 |
| V0.036W0.04Ti |
25.9 |
0.477 |
29.00 |
3.4. XRD results
To investigate the crystal structure of the active phase in the different catalysts, XRD patterns of the catalysts are shown in Fig. 3. For all catalysts, anatase phase (PDF-ICDD21-1272) was the main phase, and only a little rutile phase (PDF-ICDD21-1276) appeared. With the increasing of V molar amount from 0 to 0.036, XRD patterns of V–W–Ti catalysts showed no significant variation. It is worthy to note that the peak ascribed to crystalline V2O5 and WO3 phase was absent in the XRD patterns even if V molar ratio increased from 0.004 to 0.02. It suggested that V and W be well dispersed. However, for V0.028W0.04Ti and V0.036W0.04Ti catalysts, a V2O5 peak appeared at ∼20.2°, which indicated that vanadium oxide species transforms from monomeric or polymeric state to a bulk phase via crystallization. Bulk (i.e., crystallized) V2O5 is more inactive than monomeric or polymeric vanadium oxide species for SCR reaction. The negligible effect of increasing V amount above 0.02 on the SCR activity is related to this change.22
 |
| | Fig. 3 X-ray diffraction patterns of V–W–Ti catalysts. | |
The average crystalline size was determined according to the diffraction peak indexed to the crystal plane by using the Scherrer formula: D = 0.89λ/(βcosθ),23 where λ is the wavelength of radiation; β is the corrected peak width at half-maximum intensity (FWHM in radians); and θ is the peak position of the main reflection. The crystalline sizes of V–W–Ti catalysts were in the range of 10–14 nm in Table 3, which revealed that these catalysts prepared by the sol–gel method possess nanometer particle sizes. In addition, the diffraction peaks of V–W–Ti catalysts slightly shifted to low-angle direction compared with W–Ti catalyst, which also suggested that vanadium ions be incorporated into the lattice of TiO2 to form uniform solid solution.24
Table 3 XRD parameters and average crystal sizes of V–W–Ti catalysts
| Catalysts |
Crystal face |
λ/nm |
2θ/(°) |
β/(°) |
D/nm |
| W0.04Ti |
101 |
0.1542 |
25.35 |
0.557 |
14.9 |
| V0.004W0.04Ti |
101 |
0.1542 |
25.309 |
0.797 |
10.3 |
| V0.012W0.04Ti |
101 |
0.1542 |
25.147 |
0.64 |
12.9 |
| V0.02W0.04Ti |
101 |
0.1542 |
25.217 |
0.607 |
13.6 |
| V0.028W0.04Ti |
101 |
0.1542 |
25.146 |
0.646 |
12.8 |
| V0.036W0.04Ti |
101 |
0.1542 |
25.157 |
0.695 |
11.8 |
3.5. Raman results
To further investigate the structures of the vanadium and tungsten species, the Raman spectra are displayed in Fig. 4. Two peaks at 956 and 802 cm−1, which can be assigned to W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode and the polymeric tungsten oxide species, respectively can be detected in W0.04Ti sample.25,26 V0.004W0.04Ti catalyst showed a broad Raman band ranged from 950 to 990 cm−1, which was attributed to the overlap of symmetrical VO and WO stretching mode under monolayer coverage.27 Since the Raman cross section of two-dimensional VO species is almost four times that of two-dimensional WO species over titania surface,28,29 the Raman band of tungsten species were likely to be overshadowed by the strong Raman band of vanadium species. Therefore, the peak shift in this range mainly resulted from the structure transformation of vanadium species. With the increasing of vanadium content to 0.02, the Raman band at 977 cm−1 attributing to two-dimensional surface vanadium species became stronger. This result indicated that the partly distorted vanadium species. For V0.028W0.04Ti and V0.036W0.04Ti samples were abundant, the Raman spectra showed a sharp and intense peak at 993 cm−1 assigned to a trace of crystalline V2O5. It was consistent with the XRD results.
O stretching mode and the polymeric tungsten oxide species, respectively can be detected in W0.04Ti sample.25,26 V0.004W0.04Ti catalyst showed a broad Raman band ranged from 950 to 990 cm−1, which was attributed to the overlap of symmetrical VO and WO stretching mode under monolayer coverage.27 Since the Raman cross section of two-dimensional VO species is almost four times that of two-dimensional WO species over titania surface,28,29 the Raman band of tungsten species were likely to be overshadowed by the strong Raman band of vanadium species. Therefore, the peak shift in this range mainly resulted from the structure transformation of vanadium species. With the increasing of vanadium content to 0.02, the Raman band at 977 cm−1 attributing to two-dimensional surface vanadium species became stronger. This result indicated that the partly distorted vanadium species. For V0.028W0.04Ti and V0.036W0.04Ti samples were abundant, the Raman spectra showed a sharp and intense peak at 993 cm−1 assigned to a trace of crystalline V2O5. It was consistent with the XRD results.
 |
| | Fig. 4 Laser Raman spectra of V–W–Ti catalysts. | |
3.6. TEM results
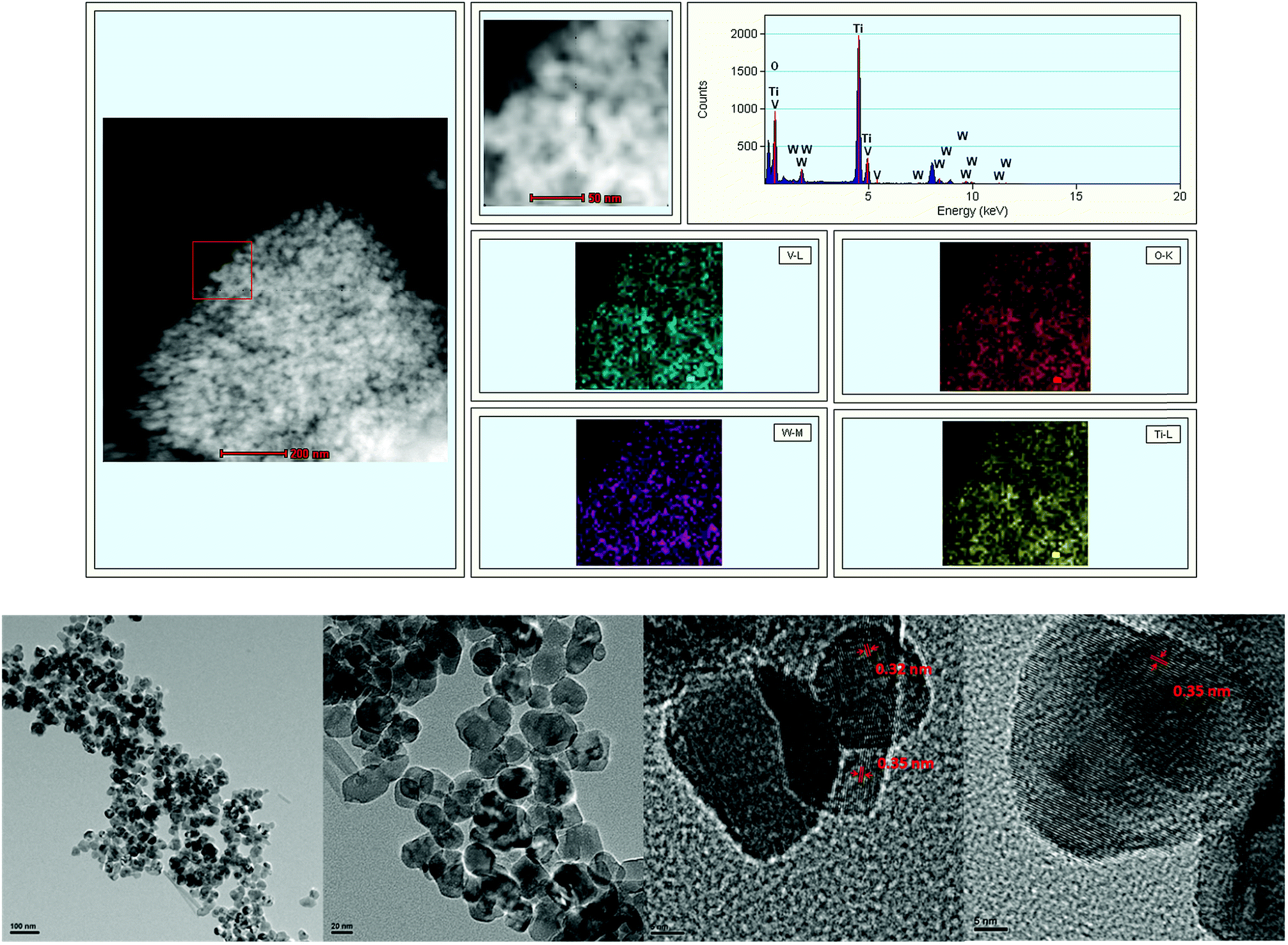
Fig. 5 shows the HAADF-TEM images of V0.02W0.04Ti catalyst together with the elemental mapping images for Ti, V, and W in the same region. In the HAADF-STEM image, small particles in the bright contrast were observed on the V0.02W0.04Ti catalyst. From the EDX mapping images, it is confirmed that tungsten elements were not aggregated, which correlates with the lack of WO3 peak in the XRD pattern. As V-L (0.511 keV) and Ti-L (0.452 keV) peaks in EDS spectrum completely overlap, they were not distinguished by current EDS spectroscopy. TiO2 grain was made of nanoparticles in the size range of 15–30 nm, as shown in Fig. 5b and c. In addition, the microstructure of the V0.02W0.04Ti catalyst was also investigated by HRTEM in Fig. 5d and e. Two phases with the interplanar spacing of 0.32 and 0.35 nm were shown in the images, which was corresponding to the interplanar distance of rutile phase (110) plane and anatase phase (101) plane, respectively. No individual particles of V2O5 and WO3 were seen on the surface of the catalyst. This result was in agreement with the XRD determination results.
 |
| | Fig. 5 HAADF-STEM images and EDS elemental mapping (a) of V0.02W0.04Ti catalyst; and TEM images of V0.02W0.04Ti catalyst with (b and c) low resolution, and (d and e) high resolution. | |
3.7. SEM results
The SEM image in Fig. 6 shows that V0.02W0.04Ti catalyst is composed of the homogeneity and monodispersity of spherical-shaped nanoparticles. The EDS results from different regions (Point 1 and Point 2) give similar compositions. The molar ratios of V/Ti and W/Ti are 0.020–0.019 and 0.035–0.036 for V0.02W0.04Ti catalyst, respectively, which are in good agreement with the initial nominal amounts.
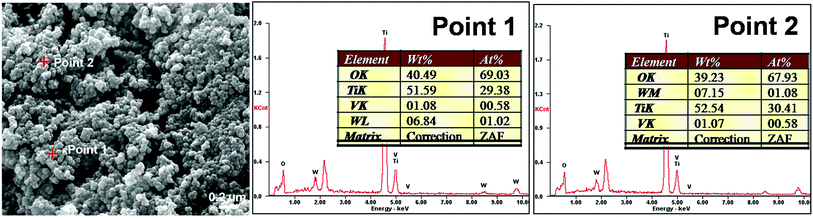 |
| | Fig. 6 SEM images and EDS analyses for V0.02W0.04Ti catalyst. | |
3.8. XPS results
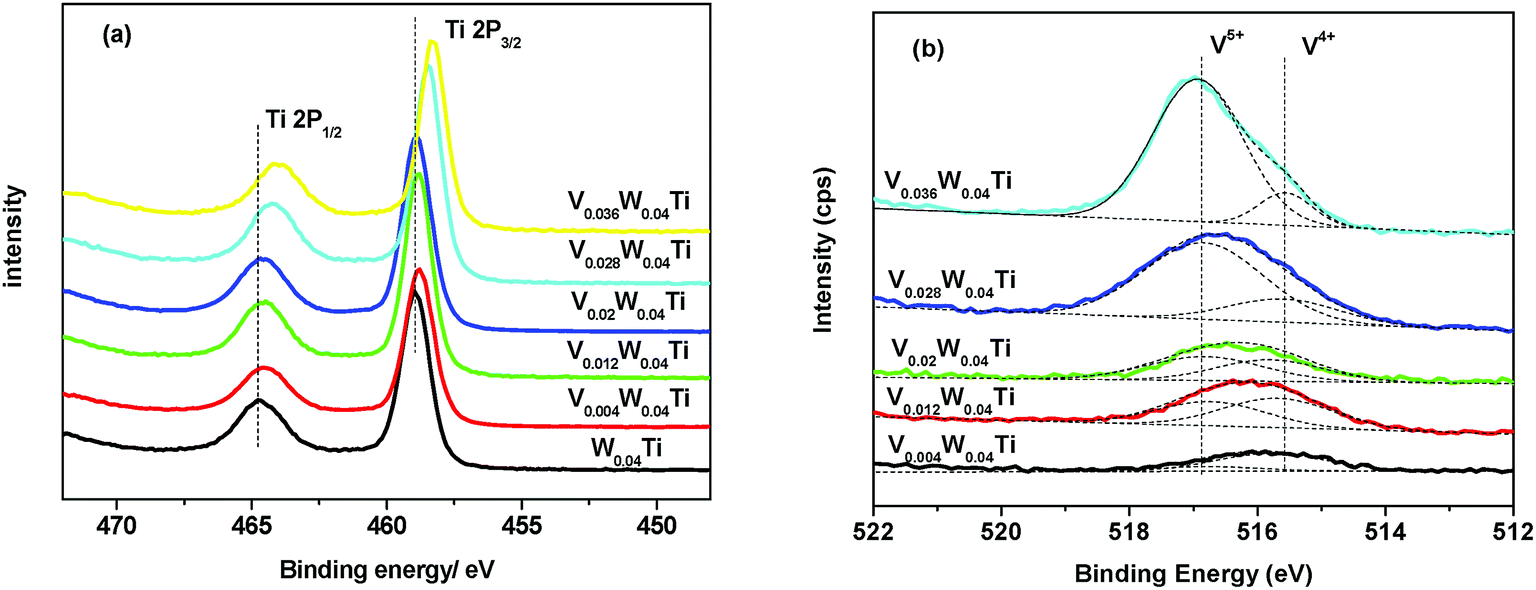
VxW0.04Ti catalysts are excellent catalysts for NH3-SCR. The XPS was carried out to investigate the chemical compositions and the valence states of the various species of the catalysts. Ti 2p XPS studies of V–W–Ti catalysts are shown in Fig. 7a. The peaks of the samples appearing at around 458.7 and 464.5 eV could be attributed to Ti 2p3/2 and Ti 2p1/2 of Ti4+, respectively.30,31 For V–W–Ti catalysts, Ti 2p position was shifted toward lower BE by 0.5 eV with increasing vanadium amount from 0 to 0.036, indicating that some Ti4+ could be reduced to Ti3+. As shown in Fig. 7b, the peaks at 516.8 and 515.6 eV could be ascribed to the 2p3/2 binding energies of V5+ and V4+.32,33 It could be seen that there is a little blue shift for V 2p3/2.
 |
| | Fig. 7 XPS spectra of (a) Ti2p and (b) V2p for various catalysts. | |
3.9. H2-TPR results
In the H2-TPR profiles of Fig. 8, it can be seen that all catalysts gave a single reduction peak at the temperature ranging from 420 to 600 °C. This peak can be assigned to the coreduction of W(VI) to W(IV) and V(V) to V(III).13 With the increasing of V doping amount from 0 to 0.02, the temperature of the reduction peak showed a monotonic decrease from 593 to 427 °C. Further increasement of V doping molar amount to 0.036 led to the higher peak temperature increase. V0.02W0.04Ti catalyst showed the lowest reduction temperature, thus it exhibited the highest reducibility. Sorrentino et al.34 reported that catalysts with good reducibility in the H2-TPR results had good catalytic activity towards the SCR process for DeNOx. The high reducibility of V0.02W0.04Ti catalyst was contributed to NH3-SCR of NO. Meanwhile, the H2 consumption of V–W–Ti catalysts were larger than that of W–Ti catalyst except for V0.004W0.04Ti, as shown in Table 4. This indicated that doping V ions into W–Ti catalyst increased the reduction degree of the catalysts, which was also correlated to the SCR activity.
 |
| | Fig. 8 H2-TPR profiles of V–W–Ti catalysts. | |
Table 4 The max temperature and area of reduction peaks in the H2-TPR
| Catalysts |
Tmaxa (°C) |
Peak area |
| The max temperature of the reduction peak. |
| W0.04Ti |
593 |
278 |
| V0.004W0.04Ti |
521 |
203 |
| V0.012W0.04Ti |
438 |
346 |
| V0.02W0.04Ti |
427 |
329 |
| V0.028W0.04Ti |
524 |
597 |
| V0.036W0.04Ti |
595 |
859 |
3.10. NH3-TPD results
Temperature-programmed desorption of ammonia was carried out to determine the strength and amount of different acid sites. The NH3-TPD patterns of V–W–Ti catalysts are depicted in Fig. 9. All catalysts showed one peak around 150–250 °C, and this peak could be ascribed to the weak acidic sites, which is corresponding to physisorbed ammonia and partially ionic NH4+ bound to Brønsted acid sites.35 A shoulder peak in the ranged 250–430 °C was detected on V0.012W0.04Ti and V0.02W0.04Ti catalysts. This peak was ascribed to the desorption of coordinated NH3 bound Lewis acid sites.36 Indeed, the ammonia adsorption ability of the catalyst is generally considered as one of the most important criteria for the screening of catalytic systems for the NH3-SCR. As was shown in Fig. 9, V0.02W0.04Ti catalyst showed predominantly more desorbed ammonia. It seems that vanadium species create new acidic sites and a progressive increasement of the surface acidity owing to the addition of vanadium. Thus, the improvement in ammonia adsorption over V0.02W0.04Ti catalyst is believed to be significantly beneficial to the NOx reduction by ammonia at the wide temperature range.
 |
| | Fig. 9 NH3-TPD curves of V–W–Ti catalysts. | |
3.11. In situ DRIFTS studies
3.11.1 Adsorption of NH3. Fig. 10 shows the in situ DRIFT spectra of NH3 adsorption over V0.02W0.04Ti catalyst at different temperatures. When NH3 was introduced into the DRIFTS cell at room temperature, several vibration bands can be detected in the range of 1000–4000 cm−1. The bands at 1632 and 1228 cm−1 can be assigned to the asymmetric and symmetric bending vibrations of NH bond in NH3 coordinately linked to Lewis acid site, respectively. And the peaks at 3348, 3256 and 3149 cm−1 can be ascribed to the N–H stretching vibration modes of the coordinated NH3, while the band at 1435 cm−1 is due to the asymmetric bending vibration of NH bond in NH4+ chemisorbed on Brønsted acid site.37,38 These adsorbed NH3 species decreased slowly with the increasing of the temperature. However it is noticeable that these vibration bands were not disappeared completely even at 350 °C. The obtained results suggested that there be both Lewis and Brønsted acid site on the surface of V0.02W0.04Ti catalyst and these adsorbed NH3 species be very stable. Previous reports showed Brønsted acid site was beneficial to the adsorption of NH3 thus improving the low-temperature activity.39,40
 |
| | Fig. 10 In situ DRIFT spectra of NH3 adsorption on V0.02W0.04Ti catalyst measured at 30–350 °C. | |
3.11.2 Co-adsorption of NO and O2. Fig. 11 shows the in situ DRIFT spectra of NO + O2 desorption on V0.02W0.04Ti catalyst at different temperatures. Several distinct bands appeared at 1241, 1292, 1580 and 1608 cm−1 at room temperature, which were respectively assigned to the asymmetric frequency of gaseous NO2 molecules (1608 cm−1), bidentate nitrate (1580 cm−1), monodentate nitrate (1292 cm−1), bridged nitrate (1241 cm−1). When the temperature was 150 °C, the bands ascribed to monodentate and bridged nitrate vanished. However, the peaks attributed to cis-N2O22− (1356 cm−1), bridged bidentate nitrite (1227 cm−1), monodentate nitrite (1443 cm−1) and chelating bidentate nitrate (1540 cm−1) appeared, suggesting a transformation from monodentate and bidentate nitrate to cis-N2O22−, bridged bidentate and monodentate nitrite over the V0.02W0.04Ti catalyst.41–44 With the increasing of the temperature to 250 °C, all bands attributed to NOx adspecies vanished, indicating that the absorbed nitrate and nitrite species were decomposed or inhibited.
 |
| | Fig. 11 In situ DRIFT spectra of NO + O2 adsorption on V0.02W0.04Ti catalyst measured at 30–350 °C. | |
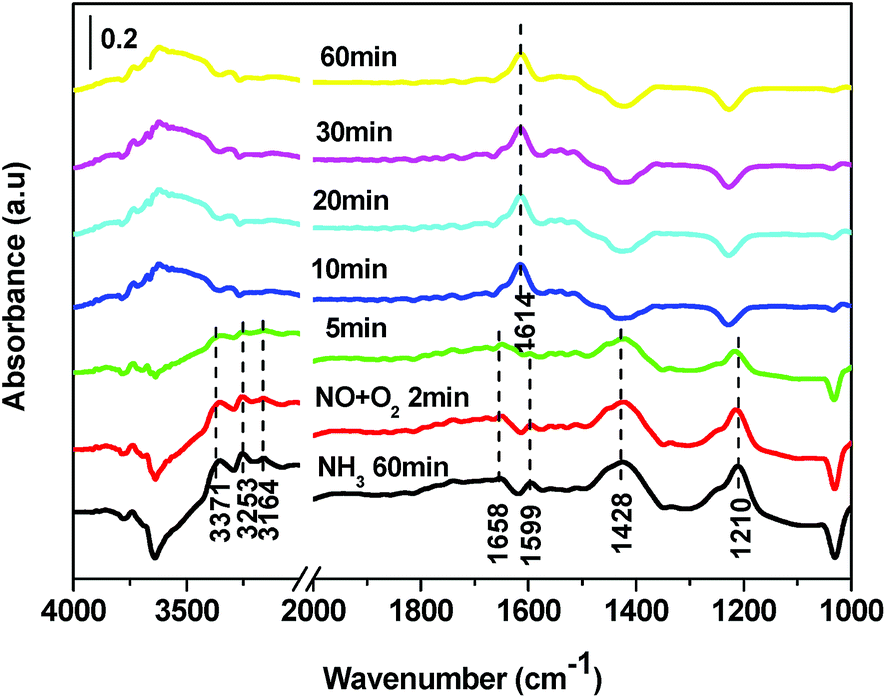
3.11.3 Reaction between nitrogen oxides and ammonia adspecies. Fig. 12 shows the in situ DRIFT spectra of V0.02W0.04Ti catalyst as a function of time in a flow of NO + O2 after the catalysts was pretreated. The coordinated NH3 on Lewis (1210 and 1599 cm−1) and Brønsted acid site (1428 and 1658 cm−1) were formed on V0.02W0.04Ti catalyst with feeding NH3. All bands due to ammonia adspecies diminished in 10 min after V0.02W0.04Ti catalyst was purged by NO + O2, and subsequently the absorbed NO2 (1614 cm−1) was observed. It could be concluded that NOx readily reacted with surface-active NH3 species over V0.02W0.04Ti catalyst. It was noticeable that the bands assigned to NOx species were absent and only one band related to NO2 appeared. This result indicated that the adsorption of nitrate species was inhibited, leading to more active sites available for the adsorption and activation of NH3. Thus a higher catalytic activity was obtained.
 |
| | Fig. 12 In situ DRIFT spectra over V0.02W0.04Ti catalyst as a function of time in a flow of NO + O2 after the catalysts was pre-exposed to a flow of NH3 for the 60 min and followed by N2 purging for 30 min at 250 °C. | |
3.11.4 Reaction between ammonia and adsorbed nitrogen oxides species. Fig. 13 shows the in situ DRIFT spectra over V0.02W0.04Ti catalyst as a function of time in a flow of NH3 after the catalysts was pre-exposed to a flow of NO + O2 for the 60 min followed by N2 purging for 30 min at 250 °C. The band for the absorbed NO2 (1614 cm−1) vanished in 5 min after NH3 was introduced into the system. Simultaneously, the peaks ascribed to coordinated NH3 on Lewis acid sites (3362, 3256, 3165, 1594, 1213 cm−1) and NH4+ species on Brønsted acid sites (1429, 1654 cm−1) appeared. All the results indicated that the absorbed NO2 species could easily react to NH4+ species to form NH4NO2 species. Thus, it may be reasonable to assume that NH4NO2 is reaction intermediate on NH3-SCR.
 |
| | Fig. 13 In situ DRIFT spectra over V0.02W0.04Ti catalyst as a function of time in a flow of NH3 after the catalysts was pre-exposed to a flow of NO + O2 for the 60 min followed by N2 purging for 30 min at 250 °C. | |
3.11.5 In situ DRIFT spectra in a flow of NO + NH3 + O2. Fig. 14 shows the in situ DRIFT spectra of V0.02W0.04Ti catalyst in a flowing of 1000 ppm NH3 + 1000 ppm NO + 3% O2 at various temperatures under the steady-state condition. No band assigned to NOx species was detected in the whole region, which was resulted from the competitive adsorption and following the reactions among NH3. With the increasing of the temperature, the intensity of the bands assigned to NH4+ species on Brønsted acid sites (1429, 1654 cm−1) decreased faster than that of the bands ascribed to coordinated NH3 on Lewis acid sites (3362, 3264, 3180, 1594, 1213 cm−1). The adsorbed NH3 species on Brønsted acid site disappeared completely at 350 °C, while the band at 1203 cm−1 attributed to the symmetric bending vibrations of N–H bond coordinately linked to Lewis acid site was still detected even at 400 °C. Combined with the results of the in situ DRIFT spectra of NH3 adsorption, Brønsted acid sites was not disappeared at 350 °C. These results indicated that the Brønsted acid site over V0.02W0.04Ti catalyst may be more active than Lewis acid site in NH3-SCR reaction.
 |
| | Fig. 14 In situ DRIFT spectra of V0.02W0.04Ti catalyst in a flowing of 1000 ppm NH3 + 1000 ppm NO + 3% O2 at 30–400 °C. | |
4. Discussion
4.1. The relation between the structures of catalysts and their catalytic performance for NH3-SCR
In the present work, V, W-incorporated TiO2 catalysts with different molar ratios were synthesized by a direct sol–gel method. When the vanadium content is no more than 0.02, the promoting effect on NH3-SCR reaction at the low temperature (below 250 °C) can be observed. Several early studies showed that the incorporation of some transition metal ions such as cerium and ferric into the framework of the support can promote remarkably the catalytic activity for NH3-SCR reaction.45 This kind of framework incorporated catalysts is different from supported catalysts. The active metal ions are fully isolated and dispersed throughout the support. Therefore, high concentrations of active sites can be achieved in framework-incorporated catalysts. Thus, this kind of catalyst exhibits a strong synergetic effect between active species and support. Similar conclusions have been reached in V–W–Ti catalysts. The XRD and XPS results demonstrate that V ions are incorporated into the framework of TiO2. V and W are well dispersed in the framework of TiO2 support and the partly distorted vanadium species are formed in V0.02W0.04Ti catalyst (Fig. 4). It is helpful to enhance the NH3-SCR activity and inhibit the vanadium loss at the high temperature. Furthermore, the vanadium oxide species was found to exist as an amorphous phase when the molar ratio of V/Ti below 0.02. Above monolayer coverage, bulk V2O5 phase appears. The highly dispersed and partly distorted vanadium species are beneficial to NH3-SCR reaction, and crystalline state V2O5 is unfavorable for this reaction.
The electronic and redox properties of V–W–Ti catalysts are also important factors for determining the extent of NO conversion in NH3-SCR reaction. The results of XPS show that there is a red shift of the binding energy of Ti 2p with the increasing of vanadium molar amount from 0.004 to 0.036 (Fig. 7). This might be caused due to the difference of ionization energy of W and V. Ion doping into the lattice would slightly change the chemical valence of Ti and destroy the charge balance of the original system. In order to maintain the charge balance in the crystal lattice, some Ti ions would release excess electrons and result in some titanium ions into a lower valence. Hence, the red-shift of Ti 2p XPS peak was observed, and Ti4+ became more electropositive, which could decrease the recombination between free electrons and holes. Importantly, the cation-anion codoping was successfully achieved by incorporating V and W into TiO2. Meanwhile, it could be seen that the peaks of Ti and V 2p3/2 of V–W–Ti catalysts shifted in the reverse direction, which was in accordance with the strong interaction between V and Ti in V–W–Ti catalysts. These results manifested that the rich-electron on the surface of the catalyst, and a higher reduction activity could be expected. The redox cycle resulted in a decrease in the energy required for the electron transfer between Ti and V active sites, promoting the activation of NH3 and NO, and consequently causing an improvement of the NH3-SCR activity. V0.02W0.04Ti catalyst showed a better balance of V4+ and V5+ in the V2p Gaussian-fitted curves than the other samples. Furthermore, the presence of more V4+ in the V0.02W0.04Ti catalyst suggests that more active sites (i.e., redox site V4+–OH) be present. Based on the above results, the redox cycle of V4+ + Ti4+ → V5+ + Ti3+ may be established, which is in agreement with the literature.42,46 The presence of mixed V4+ and V5+ enhanced the electrical conductivity.47 V0.02W0.04Ti catalyst is even more reducible than the other catalyst, as shown in H2-TPR results. Thus, it exhibits the highest catalytic activity.
4.2. The kinetic investigation and NH3-SCR mechanism on V–W–Ti catalysts
Fig. 2 shows the Arrhenius plots based on the reaction rate data between 100 and 175 °C. The A and Ea were determined from these plots given in Table 1. On V0.004W0.04Ti catalyst, A and Ea were the highest comparing to the other catalysts. There were small difference for Ea (42.4–44.1 kJ mol−1) and A (1.2–2.2 × 106) of V0.012–0.028W0.04Ti, and V0.02W0.04Ti catalyst exhibited the lowest Ea value of 42.4 kJ mol−1. It indicated that less energy was needed for activation in the reaction. Therefore, the catalytic activity was significantly improved and NO conversion would increase. Further increasing V doping molar amount to 0.036, A and Ea values increased again. This phenomenon could be explained by the change of V active sites. When the vanadium doping amount is low, V active species may be not sufficient. Then the vanadium species reach the highest dispersity with the increasing of V doping molar amount to 0.028. Further increasing vanadium molar amount to 0.036 leads to the formation of crystalline V2O5, which is harmful to the activity of NH3-SCR reaction. These results confirm the influence of vanadium oxide structure on SCR activity.
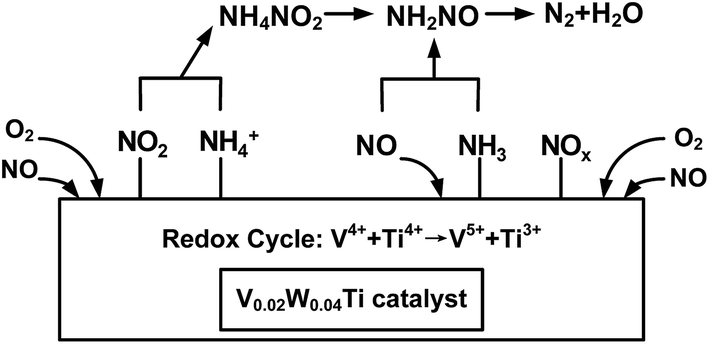
The inactive nitrate species are often thought to be thermally stable and might be accumulated with the increasing of reaction time.48 These NO3− species covered the surface active sites to prohibit the adsorption of NH3, thus showing harmful effects for NH3-SCR reaction. However, NO2 is considered to be an important intermediate species for the NH3-SCR reaction.36 In the present study, the nitrate species were highly inhibited and the adsorbed NO2 gaseous molecule was easily formed over V0.02W0.04Ti catalyst (see Fig. 11 and 12), which resulted in the NH3-SCR performance improvement. It may be attributed to the well balance V5+/V4+ present over V0.02W0.04Ti catalyst, which would promote the adsorption and activation of gas-phase oxygen. The activated oxygen atom is beneficial to the formation of NO2. From Fig. 11 and 13, it is seen that there are both Lewis and Brønsted acid site over V0.02W0.04Ti catalyst and both types of acid sites are involved in NH3-SCR reaction. It indicated that surface acidity plays an important role in the adsorption and activation of NH3, and thus affects the occurrence of SCR reaction. Based the above results and analyses, a cycle of the catalytic active site and NH3-SCR reaction mechanism is proposed over framework-incorporation V–W–Ti oxide catalysts. As shown in Fig. 15, NO2 could react with NH4+ to NH4NO2 species, which reacted further to yield the NH2NO species. Then the NH2NO decomposed to form N2 and H2O. The whole process follows the L–H mechanism. On the other hand, the adsorbed NH3 on Lewis acid sites would react with gas-phase NO (Eley–Rideal mechanism) to form N2. The catalytic active site over V0.02W0.04Ti catalyst consists of three steps. Firstly, gas-phase NH3 were absorbed on the Brønsted (V–OH) and Lewis (VO) acid sites. Secondly, these adsorbed NH3 species reacted with absorbed NO2 or gas-phase NO and V5+O were reduced to V4+–OH. Finally, V4+–OH were re-oxidized to V5+O by O2 and the catalyst was regenerated. Therefore, the redox couples of V4+/V5+ play an important role for the SCR reaction to proceed. The existence of redox cycle (V4+ + Ti4+ → V5+ + Ti3+) can contribute to the redox process of V4+/V5+ and an improvement of the NH3-SCR activity.
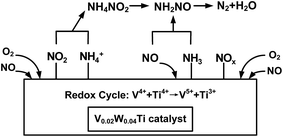 |
| | Fig. 15 Schematic diagram of NH3-SCR in the presence of excess oxygen over V0.02W0.04Ti catalyst. | |
5. Conclusion
Novel framework-incorporation V–W–Ti nanoparticle catalysts with different vanadium contents for the selective catalytic reduction of NOx have been developed by direct sol–gel method. V0.02W0.04Ti catalyst exhibited the high NO conversion (>90%) in the wide temperature range (200–400 °C) and the lowest Ea value.
Vanadium was found to incorporate into the framework of TiO2 and existed as an amorphous phase when the molar ratio of V/Ti is ≤0.02. V0.02W0.04Ti catalyst with the best vanadium dispersion exhibited the best catalytic performance for the NH3-SCR reaction. With the increasing of vanadium doping amount, vanadium species were transformed from partly distorted vanadium species to crystalline V2O5. The redox cycle (V4+ + Ti4+ → V5+ + Ti3+) and the well balance V5+/V4+ play key roles for the highly catalytic deNOx performance.
The reaction mechanism for NH3-SCR over V0.02W0.04Ti catalyst was studied by means of in situ DRIFT spectroscopy. The results demonstrated that Brønsted acid site over V0.02W0.04Ti catalyst were more active in NH3-SCR reaction. On the one hand, the catalyst prevented the formation of stable nitrate, leaving more active sites available for the absorption of NH3. On the other hand, the adsorbed NO2 gaseous molecule was easily formed on the catalyst surface to promote the activity of V0.02W0.04Ti catalyst.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21376261 and 21173270), 863 Program (2015AA034603 and 2013AA065302), the Beijing Natural Science Foundation (2142027), and the China University of Doctoral Selective Fund (20130007110007).
References
- J.-O. Barth, A. Jentys and J. A. Lercher, Ind. Eng. Chem. Res., 2004, 43, 3097 CrossRef CAS.
- T. Zhang, J. Liu, D. Wang, Z. Zhao, Y. Wei, K. Cheng, G. Jiang and A. Duan, Appl. Catal., B, 2014, 148–149, 520 CrossRef CAS PubMed.
- G. Busca, L. Lietti, G. Ramis and F. Berti, Appl. Catal., B, 1998, 18, 1 CrossRef CAS.
- P. Forzatti, Appl. Catal., A, 2001, 222, 221 CrossRef CAS.
- S. Roy, M. S. Hegde and G. Madras, Appl. Energy, 2009, 86, 2283 CrossRef CAS PubMed.
- L. Dong, C. Sun, C. Tang, B. Zhang, J. Zhu, B. Liu, F. Gao, Y. Hu, L. Dong and Y. Chen, Appl. Catal., A, 2012, 431–432, 126 CrossRef CAS PubMed.
- I. E. Wachs and C. A. Roberts, Chem. Soc. Rev., 2010, 39, 5002 RSC.
- A. Khodakov, B. Olthof, A. T. Bell and E. Iglesia, J. Catal., 1999, 181, 205 CrossRef CAS.
- G. T. Went, S. T. Oyama and A. T. Bell, J. Phys. Chem., 1990, 94, 4240 CrossRef CAS.
- I. Giakoumelou, C. Fountzoula, C. Kordulis and S. Boghosian, J. Catal., 2006, 239, 1 CrossRef CAS PubMed.
- R. Willi, M. Maciejewski, U. Gobel, R. A. Koppel and A. Baiker, J. Catal., 1997, 166, 356 CrossRef CAS.
- T. S. Park, S. K. Jeong, S. H. Hong and S. C. Hong, Ind. Eng. Chem. Res., 2001, 40, 4491 CrossRef CAS.
- G. Busca, G. Centi, L. Marchetti and F. Trifiro, Langmuir, 1986, 2, 568 CrossRef CAS.
- W. Shan, F. Liu, H. He, X. Shi and C. Zhang, Appl. Catal., B, 2012, 115–116, 100 CrossRef CAS PubMed.
- Y. Peng, K. Li and J. Li, Appl. Catal., B, 2013, 140–141, 483 CrossRef CAS PubMed.
- G. Cristallo, E. Roncari, A. Rinaldo and F. Trifirò, Appl. Catal., A, 2001, 209, 249 CrossRef CAS.
- P. G. W. A. Kompio, A. Brückner, F. Hipler, G. Auer, E. Löffler and W. Grünert, J. Catal., 2012, 286, 237 CrossRef CAS PubMed.
- H. Y. Huang, R. Q. Long and R. T. Yang, Appl. Catal., A, 2002, 235, 241 CrossRef CAS.
- F. Gao, E. D. Walter, E. M. Karp, J. Luo, R. G. Tonkyn, J. H. Kwak, J. Szanyi and C. H. F. Peden, J. Catal., 2013, 300, 20 CrossRef CAS PubMed.
- P. Ciambelli, M. E. Fortuna, D. Sannino and A. Baldacci, Catal. Today, 1996, 29, 161 CrossRef CAS.
- H. S. Fogler, Elements of chemical reaction engineering, Prentice Hall PTR, India, 3rd edn, 1997 Search PubMed.
- S. Chin, E. Park, M. Kim, G. N. Bae and J. Jurng, J. Colloid Interface Sci., 2011, 362, 470 CrossRef CAS PubMed.
- H. Klug and L. Alexander, X-ray Diffraction Procedures for Poly-crystalline and Amorphous Materials, John Wiley, New York, 2nd edn, 1974 Search PubMed.
- X. Yao, L. Zhang, L. Li, L. Liu, Y. Cao, X. Dong, F. Gao, Y. Deng, C. Tang, Z. Chen, L. Dong and Y. Chen, Appl. Catal., B, 2014, 150–151, 315–329 CrossRef CAS PubMed.
- L. J. Alemany, L. Lietti, N. Ferlazzo and P. Forzatti, J. Catal., 1995, 155, 117 CrossRef CAS.
- C. Su, L. Dong, W. Yu, L. Liu, L. Hao, F. Gao, L. Dong and Y. Chen, J. Mol. Catal. A: Chem., 2011, 346, 29 CrossRef PubMed.
- M. A. Reiche, T. Bürgi, A. Baiker, A. Scholz, B. Schnyder and A. Wokaun, Appl. Catal., A, 2000, 198, 155 CrossRef CAS.
- M. A. Vuurman, I. E. Wachs and A. M. Hirt, J. Phys. Chem., 1991, 95, 9928 CrossRef CAS.
- C. Wang, S. Yang, H. Chang, Y. Peng and J. Li, Chem. Eng. J., 2013, 225, 520 CrossRef CAS PubMed.
- B. M. Reddy, A. Khan, Y. Yamada, T. Kobayashi, S. Loridant and J. Volta, J. Phys. Chem. B, 2003, 107, 5162 CrossRef CAS.
- K. Cheng, J. Liu, T. Zhang, J. Li, Z. Zhao, Y. Wei, G. Jiang and A. Duan, J. Environ. Sci., 2014, 26, 2106 CrossRef PubMed.
- V. I. Bukhtiyarov, XPS and SIMS characterization, Catal. Today, 2000, 56, 403 CrossRef CAS.
- L. K. Boudali, A. Ghorbel, P. Grange and F. Figueras, Appl. Catal., B, 2005, 59, 105 CrossRef CAS PubMed.
- A. Sorrentino, S. Regaa, D. Sannino, A. Magliano, P. Ciambelli and E. Santacesaria, Appl. Catal., A, 2001, 209, 45 CrossRef CAS.
- K. J. Lee, P. A. Kumar, M. S. Maqbool, K. N. Rao, K. H. Song and H. P. Ha, Appl. Catal., B, 2013, 142, 705 CrossRef PubMed.
- L. Chmielarz, R. Dziembaj, T. Grzybek, J. Klinik, T. Lojewski, D. Olszewska and A. Wegrzyn, Catal. Lett., 2000, 70, 51 CrossRef CAS.
- L. Lietti, I. Nova, G. Ramis, L. DalllAcqua, G. Busca, E. Giamello, P. Forzatti and F. Bregani, J. Catal., 1999, 187, 419 CrossRef CAS.
- P. G. Smirniotis, D. A. Peña and B. S. Uphade, Angew. Chem., Int. Ed., 2001, 40, 2479 CrossRef CAS.
- L. Chen, J. Li and M. Ge, Environ. Sci. Technol., 2010, 44, 9590 CrossRef CAS PubMed.
- G. Zhou, B. Zhong, W. Wang, X. Guan, B. Huang, D. Ye and H. Wu, Catal. Today, 2011, 175, 157 CrossRef CAS PubMed.
- Z. Liu, J. Zhu, J. Li, L. Ma and S. I. Woo, ACS Appl. Mater. Interfaces, 2014, 6, 14500 CAS.
- Z. Liu, S. Zhang, J. Li, J. Zhu and L. Ma, Appl. Catal., B, 2014, 158–159, 11 CrossRef CAS PubMed.
- Z. Wu, B. Jiang, Y. Liu, H. Wang and R. Jin, Environ. Sci. Technol., 2007, 41, 5812 CrossRef CAS.
- G. Qi, R. T. Yang and R. Chang, Appl. Catal., B, 2004, 51, 93 CrossRef CAS PubMed.
- P. Li, Y. Xin, Q. Li, Z. Wang, Z. Zhang and L. Zheng, Environ. Sci. Technol., 2012, 46, 9600 CrossRef CAS PubMed.
- F. Liu, H. He, C. Zhang, W. Shan and X. Shi, Catal. Today, 2011, 175, 18 CrossRef CAS PubMed.
- W. Zhao, Q. Zhong, Y. Pan and R. Zhang, Chem. Eng. J., 2013, 228, 815 CrossRef CAS PubMed.
- L. Sun, Q. Cao, B. Hu, J. Li, J. Hao, G. Jing and X. Tang, Appl. Catal., A, 2011, 393, 323 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.