DOI:
10.1039/C5RA02837J
(Paper)
RSC Adv., 2015,
5, 45184-45193
Glycerol steam reforming over La–Ce–Co mixed oxide-derived cobalt catalysts
Received
13th February 2015
, Accepted 8th May 2015
First published on 8th May 2015
Abstract
La–Ce–Co mixed oxide derived Co catalysts were prepared by a co-precipitation method varying the molar ratio of Ce/La in the range of 0.1–0.9, but keeping the Co content constant (1 mol). The physico-chemical properties of the samples were investigated by Atomic Absorption Spectrometer (AAS), X-ray diffraction (XRD), laser Raman spectroscopy, scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), UV-diffused reflectance spectroscopy, BET surface area, hydrogen chemisorption and temperature programmed reduction techniques. These catalysts, after reduction, were evaluated for glycerol steam reforming in the temperature range of 500–700 °C and at atmospheric pressure. The characterization results of the catalysts after reduction revealed the collapse of the perovskite structure and dispersion of Co-metal on the mixed oxide (La2O3–CeO2) support. The possibility for tuning the dispersion of the Co metal to derive maximum activity and minimum coke formation was examined. The catalyst derived from the mixed oxide, La0.7Ce0.3CoO3, exhibited the best catalytic activity at 700 °C with complete conversion of glycerol and a 68% hydrogen yield. The active cobalt area contributes a major part of the BET surface area of the reduced catalysts, and this is correlated well with the turnover frequency (TOF). The high dispersion with the formation of Co particles of 4–5 nm size, helped in achieving low coke formation.
1. Introduction
After realizing that global warming is majorly caused by burning of fossil fuels, the search for renewable feedstocks has been intensified to meet the energy requirements of the world. Biomass conversion to hydrogen has thus acquired greater prominence. The advantage of using hydrogen in fuel cells has further enhanced its significance in the energy conversion and management sector.1 Glycerol, which is obtained as a by-product in biodiesel production, can be an important source for hydrogen generation. More and more crude glycerol would be available as the production of biodiesel increases. The direct use of crude glycerol in food, health care and pharmaceutical industry is not possible due to the presence of unreacted methanol, catalyst residues and water as impurities. Since purification is cumbersome and expensive, proper utilization of crude glycerol becomes an important issue for improving the economics of biodiesel production. An efficient application of crude glycerol is to generate hydrogen by steam reforming, thus affording clean energy2–5 made available to the fuel industry.
Among the different reforming processes for glycerol conversion to hydrogen, aqueous phase reforming (APR)6–9 and steam reforming (SR)10–13 are well investigated. APR is a pressure process and offers lower hydrogen selectivity. SR, on the other hand, is favourable even at atmospheric pressure. However, the endothermic nature of SR demands high reaction temperatures leading to the formation of a variety of products and severe coking of catalyst. Thus, the design of catalyst becomes a challenging task.
The reactions associated with steam reforming process are summarized below.
| C3H8O3 + 3H2O → 7H2 + 3CO2 – steam reforming |
| C3H8O3 → 4H2 + 3CO – direct decomposition |
| CO + H2O → H2 + CO2 – WGS reaction |
| CO + 3H2 → CH4 + H2O – methanation |
| CH4 + CO2 → 2H2 + 2CO – methane dry reforming |
| 2CO → CO2 + C – carbon formation |
Pd,14 Ir, Co,15 Rh, Ru,14 Pt14,16 and Ni14,15,17,18 are investigated as catalysts for the steam reforming reaction. The noble metals are considered economically less viable because of their low availability and high cost. Though, Ni is cheaper compared to the noble metals it is associated with severe coking under reforming conditions. Extensive work has been reported in the literature to make Ni catalysts more adoptive, especially for the steam reforming of glycerol.19,20 Recent review on glycerol steam reforming illustrating the several challenging issues in producing the highly pure hydrogen.21 The new concept like continuous sorption-enhanced steam reforming by moving-beds and riser systems,19,21 sorption enhanced chemical looping was efficient methods and offering the good stability for the catalysts. Highly dispersed Ni particles are recommended for suppressing the carbon formation to a great extent.22 The attainment of such small particles depends on factors like the method of preparation, type of precursor used in the preparation,23 calcination and reaction temperatures.24 Replacement of the conventional catalysts with well defined structures like hydrotalcites, and modification of the support and active metal with suitable promoters25 are reported. CeO2 modification of Al2O3 is stated to have stabilized the Ni0 particles by enhancing nickel–ceria interactions. However, higher ceria content reduces the capacity of the Ni catalyst to convert intermediate oxygenated hydrocarbons into H2. Ni catalysts with basic oxide supports (Ex. dolomite, MgO) afford a much higher conversion of glycerol and the selectivity towards hydrogen than the acidic oxide supports (Ex. SiO2, Al2O3).26 Molybdate modification is found to decrease the acidity and the strong metal-support interaction in Ni/γ-Al2O3 catalyst leading to a stable catalytic activity.27 The substitution of 50% La with Ce in LaNiO3 mixed oxide has provided more deactivation-resistant catalyst, through the CeO2–La2O3 solid solution formation.28 Synergetic interaction of La and Ca in La0.5Ca0.5NiO3 is found to increase the metal dispersion and minimize coke formation over the Ni sites.29 The addition of small amount of lanthana (3 wt%) to Al2O3 support is advantageously adopted to improve metal dispersion and decrease Ni particle size.30 Similarly, the inclusion of CaO to ZrO2 is shown to enhance the reforming activity of Ni by minimizing the carbon formation.31 Pt is a good promoter to increase the reducibility of Ni catalysts through the H2 spillover. The side reactions are prevented, thereby increasing the catalytic activity.32 Cu insertion into the LaNiO3 perovskite has minimized the sintering of Ni particle offering good stability to the catalyst.33
Though Co based catalysts are more resistant to carbon deposition than the Ni based ones, detailed work as described above, has not been done in the identification of the influence of particle size on the activity and coke resistance in steam reforming of glycerol. Coke formation in steam reforming can be minimized by fixing the active metal in a well-defined structure like perovskite.34 These structures enhance the activity and stability,35 because they lead to nanosized particles of active metal well dispersed on the supports,36 during the course of pre-reduction. La and Co containing perovskites have been widely studied for oxidation reactions, with the incorporation of Ce in it. Ce sets up redox couple under the reaction conditions and it also possesses high oxygen storage capacity. These characteristics of the catalyst have been exploited in CO oxidation. In the case of WGS reaction it is used to maximize hydrogen production.37–41 La2O3 on the other hand, imparts stability to the catalyst.42,43 Co in combination with La and Ce would be advantageous for the maximization of hydrogen selectivity. The aim of this work is to tune Co dispersion in order to arrive at maximum hydrogen yield and minimum coke formation during the reforming reaction. La–Ce–Co mixed oxides, with varying La/Ce ratio, are selected as precursors.
2. Experimental
2.1 Catalyst preparation
The LaxCe1−xCoO3 (x = 0.1, 0.3, 0.5, 0.7, 0.9) mixed oxides were prepared by co-precipitation method. Required quantities of the nitrate salts of the metals [La(NO3)3·6H2O, Ce(NO3)3·6H2O and Co(NO3)2·6H2O (99.0%, Sigma Aldrich)] were dissolved in water and the precipitation was carried out using 5% aq. NH4OH with rigorous stirring, maintaining the pH at 10. The resulting precipitate was filtered, washed with distilled water and dried at 100 °C for 24 h. The solid mass was then carefully ground in a mortar and finally calcined at 700 °C for 6 h. The cobalt content was fixed such that the finished catalysts contained 1 mol of Co in the mixed oxides.
2.2 Characterization techniques
BET surface areas were determined by N2 adsorption on a SMART SORB 92/93 instrument (Ms Smart Instruments, India). Prior to the measurement, the samples were dried at 150 °C for 2 h. X-ray diffraction (XRD) patterns of the catalysts were obtained on an Ultima-IV diffractometer (M/s. Rigaku Corporation, Japan) using nickel-filtered Cu Kα radiation (λ = 1.54 Å). The nature of the phase in the sample was checked using the data base of the Joint Committee on Powder Diffraction Standards (JCPDS). FT-IR spectra were recorded on a Biorad-Excalibur series (USA) spectrometer using the KBr disc method. Temperature programmed reduction (TPR) of the sample was performed in a flow of 5% H2–Ar gas mixture flowing at a rate of 30 mL min−1 with a temperature ramp of 10 °C min−1. Prior to the TPR run, the sample (20 mg) was pretreated with Ar at 200 °C for 2 h. The hydrogen consumption was monitored by using the thermal conductivity detector (TCD) of a gas chromatograph. Scanning electron microscopic pictures of the catalysts were obtained on a S-520 electron microscope (M/s. Hitachi Japan) running at an accelerated voltage of 10 kV. The samples were mounted on aluminum stubs using double-adhesive tape and gold coated in a Hitachi HUS-5GB vacuum evaporator. The Raman spectra of the samples were collected on a Horiba-Jobin Yvon LabRam-HR spectrometer equipped with a confocal microscope, 2400/900 grooves/mm gratings, and a notch filter. The visible laser excitation at 532 nm (visible/green) was used. The scattered photons were directed and focused onto a single-stage monochromator and measured with a UV-sensitive LN2-cooled CCD detector.). XPS measurements were conducted on a KRATOS AXIS 165 with a dual anode (Mg and Al) apparatus using Mg Kα anode. The non-monochromatized Al Kα X-ray source (hυ) (1486.6 eV) was operated at 12.5 kV and 16 mA. Before acquisition of the data, the samples were out-gassed for 3 h at 100 °C under a vacuum of 1.0 × 10−7 torr to minimize surface contamination. The XPS instrument was calibrated using Au as standard. For energy calibration, the carbon 1s photoelectron line was used. The carbon 1s binding energy 285 eV was taken as reference. A charge neutralization of 2 eV was used to balance the charge up of the sample. The spectra were deconvoluted using a Sun Solaris based Vision-2 curve resolver. The location and the full width at half-maximum (FWHM) value for the species were first determined using the spectrum of the pure sample. Symmetric Gaussian shapes were used in all cases. UV-Vis DRS spectra were recorded in the region of 200–800 nm at a split width of 1.5 nm and scan speed of 400 nm min−1 with GBC Cintra 10e spectrometer. Pellets were made from the solid mixture contaianing 15 mg of the sample and required quantity of dried KBr, ground thoroughly for uniform mixing. The spectra were recorded at room temperature. H2 Chemisorption studies were performed using an Autosorb iQ (Quantachrome USA) unit. Catalyst samples (100 mg), taken in a quartz reactor, were first reduced in H2 gas at a flow rate of 60 mL min−1 and with a heating rate of 10 °C min−1 up to 650 °C. The samples were then flushed with He for 1 h followed by cooling. H2 gas was then introduced in pulses and the adsorption uptake was analyzed using the thermal conductivity detector of a gas chromatograph. The coke content of the used catalysts was determined in a CHNS analyzer (ElementaV, Germany). Elemental analysis was carried out to know the composition of the mixed oxides using the Atomic Absorption Spectrometer (AAS) (M/s. ANALYST-300, PERKIN-ELMAR, USA). BET, XRD, FT-IR, laser Raman, and UV-DRS characterizations were also carried out on the reduced catalysts to investigate the structural changes during reduction. The catalysts were reduced with H2 (30 mL min−1)at 650 °C/5 h cooled to the room temperature and passivated with N2 flow, before the analysis.
2.3 Reforming activity
Steam reforming of glycerol was performed in a fixed bed reactor. About 1 g of the catalyst was loaded in the middle of the reactor, suspended between two quartz wool plugs. Prior to the reaction, the catalyst was reduced with pure hydrogen (100 mL min−1) for 5 h at 650 °C. After reduction, the hydrogen gas was swept away with N2 and the reactor bed temperature was brought to the required level under N2 flow. An aqueous solution of 30 wt% glycerol was introduced into the preheater, kept above the reactor, at a rate of 0.08 mL h−1 using a HPLC pump (LabAlliance). After reaching the steady state, over a period of 1 h, the product mixture coming out of the reactor was condensed, the liquid products seperated in a gas–liquid seperator and the gas products analyzed, on-line using a gas chromatograph (Agilent,7820A) equipped with a carbosieve packed column for quantifying H2, CO, CH4 and CO2. The liquid products (acetone, hydroxy acetone, methanol, ethanol and 1,2- and 1,3-propanediol) and unreacted glycerol were analyzed by gas chromatograph using an innowax capillary column. The data acquisition was done using the following equations.
R is the H2/CO2 reforming ratio (7/3) for glycerol.
where i is CO, CH4 and CO2 produced experimentally.
3. Results and discussion
3.1 AAS, BET surface area and X-ray diffraction
The composition of the catalysts, determined by the AAS (Table 1) clearly matches with those of theoretical values. In all the catalysts, the AAS results clearly indicate that the variation of La/Ce with fixed Co loading is in accordance with the catalyst composition.
Table 1 Catalysts composition determined from AASa
| Catalysts |
La |
Ce |
Co |
| a |
b |
a |
b |
a |
b |
| a = wt%, b = molar ratio. |
| La0.9Ce0.1CoO3 |
64.23 |
0.895 |
06.94 |
0.097 |
28.83 |
0.955 |
| La0.7Ce0.3CoO3 |
49.50 |
0.699 |
20.78 |
0.296 |
29.72 |
0.998 |
| La0.5Ce0.5CoO3 |
35.67 |
0.499 |
35.22 |
0.497 |
29.11 |
0.968 |
| La0.3Ce0.7CoO3 |
20.12 |
0.277 |
50.10 |
0.698 |
29.78 |
0.978 |
| La0.1Ce0.9CoO3 |
06.90 |
0.097 |
63.27 |
0.898 |
29.83 |
0.998 |
BET area values of the samples are presented in the Table 2. The surface area of the fresh catalysts dropped from 20.1 to 8.5 m2 g−1 with increasing La content. However, after reduction in hydrogen, the surface area increased with increasing La content upto catalyst La0.7Ce0.3CoO3 and then on it decreased. The decrease in the case of the fresh catalysts may be due to perovskite formation as reported in the literature.44 In the case of the reduced samples the increase in surface area can be associated with the collapse of the structure and formation on a well dispersed Co on oxide supports.
Table 2 BET surface area, H2 chemisorption and CHNS data
| Catalyst |
BET surface area fresh samples (m2 g−1) |
BET surface area reduced samples (m2 g−1) |
Active metal surface areaa (m2 g−1) |
Average crystalliteb size (nm) |
Co dispersionc (%) |
Carbon content (wt%) |
Active metal surface area (ASA) = (Nm × S × Am)/166 where ASA is in m2 per gram of sample.  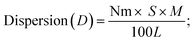 Nm = the number of adsorbed gas molecules, S = adsorption stoichiometry, Am = the cross-sectional area occupied by each active metal surface atom, M and L are the molecular weight and percent loading of the supported metal, f is a particle shape correction factor (=6 for spherical particles). Z = density of the supported metal. Nm = the number of adsorbed gas molecules, S = adsorption stoichiometry, Am = the cross-sectional area occupied by each active metal surface atom, M and L are the molecular weight and percent loading of the supported metal, f is a particle shape correction factor (=6 for spherical particles). Z = density of the supported metal. |
| La0.1Ce0.9CoO3 |
20.1 |
23.8 |
20.8 |
9.7 |
10.3 |
3.7 |
| La0.3Ce0.7CoO3 |
16.8 |
38.0 |
31.6 |
6.4 |
15.6 |
2.8 |
| La0.5Ce0.5CoO3 |
12.6 |
43.4 |
40.7 |
5.0 |
20.1 |
1.7 |
| La0.7Ce0.3CoO3 |
11.3 |
58.0 |
45.2 |
4.5 |
22.2 |
0.7 |
| La0.9Ce0.1CoO3 |
8.5 |
19.0 |
15.0 |
13.5 |
7.4 |
2.4 |
The XRD patterns of the calcined catalysts are displayed in Fig. 1(A). The peaks appearing at 36.0, 43.7 and 64.0° are assignable to the cubic Co3O4 [PCPDF-801541]. The prominent reflections for sample (a) appearing at 28.5, 33.0, 47.4, 56 and 69.4° are characteristic of CeO2 in its fluorite structure [PCPDF-710567]. Thus, samples from (a) to (c) contain predominantly Co3O4 and CeO2. In the case of samples (d) and (e), the peaks observed at 2θ values of 32.6, 46.7, 58.3 and 76.9° indicate the presence of the characteristic rhombohedral LaCoO3 perovskite [PCPDF-861665]. The peaks due to Co3O4 dominated the patterns of low La containing samples, whereas catalyst (e) contained no Co3O4. With increasing CeO2, the substitution of Ce for La seems to have increased, as reflected by the broadening of the lines.
 |
| | Fig. 1 XRD patterns of (A) calcined and (B) reduced catalysts. (a) La0.1Ce0.9CoO3 (b) La0.3Ce0.7CoO3 (c) La0.5Ce0.5Co O3 (d) La0.7Ce0.3CoO3 (e) La0.9Ce0.1CoO3. | |
The XRD patterns of reduced samples are displayed in Fig. 1(B). The peaks positioned at 2θ values of 44.1, 51.6 51.7, 55.9 and 75.6° are assigned to the metallic Co particles.45–48 The peak observed at 28.5° can be assigned to the CeO2 phase [PCPDF-710567]. CeO2 undergoes partial reduction during hydrogen treatment. However, the partially reduced phase either remained amorphous or was reoxidized (when exposed to air during the analysis). This indicates that the perovskite structure is destroyed during the reduction process.47
3.2 Fourier transform infrared spectroscopy
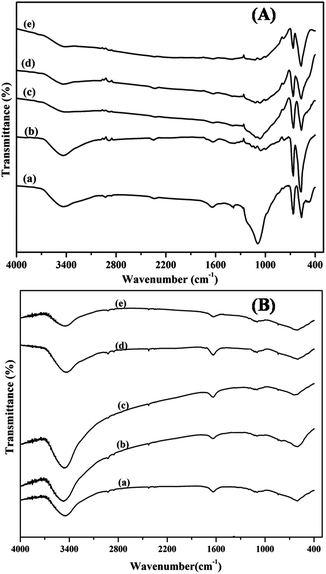
The FT-IR spectra of the calcined mixed oxide samples are shown in Fig. 2(A). The spectra display two distinctive bands originating from the stretching vibrations of the M–O bond. The first band at 570 cm−1 is associated with the vibrations in the spinel lattice, where the Co cations exist in an octahedral position and the second band at 661 cm−1 is attributed to the LaCoO3 vibrations. The Co/Ce peak intensity ratio decreased with increasing La content, as also reported in the literature.49 The peak at 3500 cm−1 can be attributed to the vibrational frequency of the –OH group of the adsorbed water. The bands at 1038, 1381 and 1633 cm−1 correspond to the vibrational frequencies of the residual ν(NO3).2–51 The spectra of the samples show an absorption band at about 1380 cm−1 which is the characteristic vibration mode of CeO2.52 The fundamental vibrational modes of La–O which normally appear between 920–940 cm−1 are not very clear.53 In the FT-IR spectra of the reduced samples shown in Fig. 2(B), the intensity of the bands at 570 and 661 cm−1 is considerably decreased indicating the reduction of cobalt oxide to its metallic state.
 |
| | Fig. 2 FT-IR spectra of (A) calcined and (B) reduced catalysts (a) La0.1Ce0.9CoO3 (b) La0.3Ce0.7CoO3 (c) La0.5Ce0.5CoO3 (d) La0.7Ce0.3CoO3 (e) La0.9Ce0.1CoO3. | |
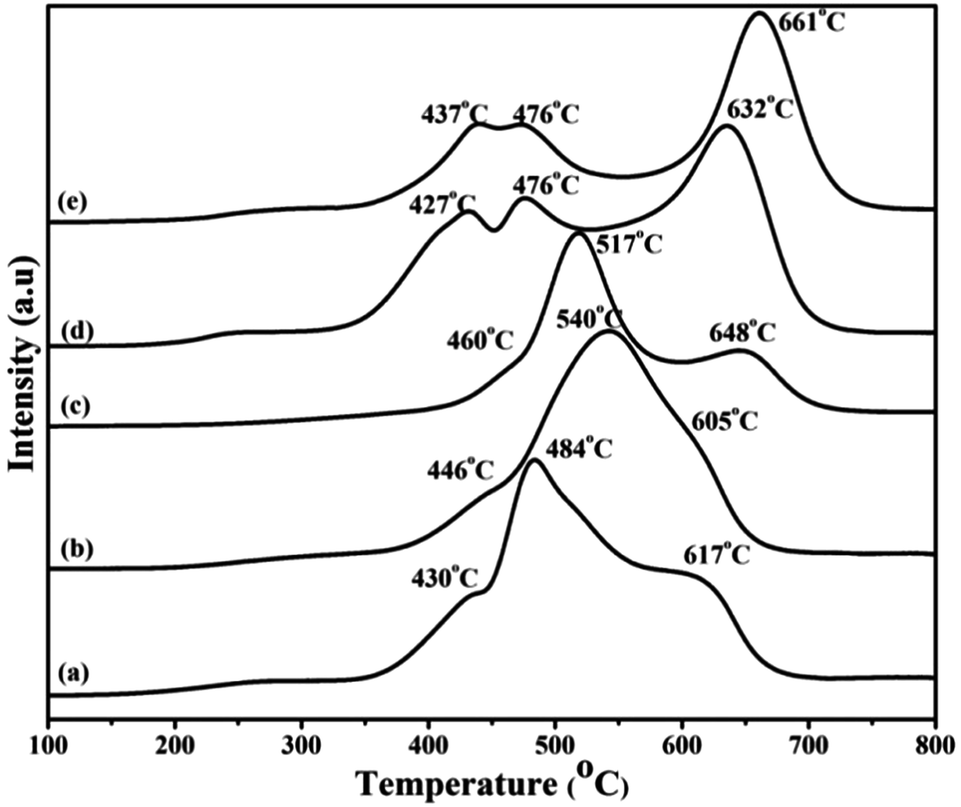
3.3 Temperature programmed reduction
The TPR patterns shown in Fig. 3 can be divided into two groups; the high Ce containing ((a) to (c)) and the low Ce containing ((d) and (e)) ones. In the case of catalysts with higher Ce content the reduction peak maxima are seen at lower temperatures due to the presence of well dispersed, small Co3O4 particles in intimate contact with CeO2 and are quite identical with the cerium supported cobalt oxide. The small hump in the beginning of the first peak at 430 °C represents the isolated cobalt oxide species and the reduction peak at 484 °C corresponds to the reduction of Co3+ to Co2+, while the second reduction peak at 617 °C represents the reduction of Co2+ to Co0. In the first three catalysts, CeO2 supports the reducibility of Co3O4. With decreasing Ce content this effect decreases. This is reflected in the positions of the peak maxima.54 The reduction profile of the LaCoO3 perovskite is different from the simple cobalt oxide supported on La2O3. In samples (d) and (e) the formation of the perovskite occurs, as shown in the XRD patterns. These TPR profiles are also similar to the ones reported in the literature.55,56 The high temperature peak is shifted to higher region. In sample (d) this peak is observed at relatively lower temperature (632 °C) than in sample (e) because of the presence of more amount of Ce in (d).
 |
| | Fig. 3 TPR of (a) La0.1Ce0.9CoO3 (b) La0.3Ce0.7CoO3 (c) La0.5Ce0.5CoO3 (d) La0.7Ce0.3CoO3 (e) La0.9Ce0.1CoO3. | |
3.4 Scanning electron microscopy
The SEM images of the fresh catalysts are shown in Fig. 4. The particle dimensions seem to have changed with composition. Samples (a) to (c) display fine distribution of mixed oxide particles. The formation and development bulk LaCoO3 particle could be seen in samples (d) and (e). The decrease in BET surface area (Table 2) of fresh samples supports this observation.
 |
| | Fig. 4 SEM images of (a) La0.1Ce0.9CoO3 (b) La0.3Ce0.7CoO3 (c) La0.5Ce0.5CoO3 (d) La0.7Ce0.3CoO3 (e) La0.9Ce0.1CoO3. | |
3.5 Laser Raman
The laser Raman spectra of the calcined and reduced samples are presented in Fig. 5(A) and (B), respectively. The five active modes A1g (679 cm−1), F2g (619, 515 and 192 cm−1) and Eg (473 cm−1) for the cobalt oxide species are observed in all the samples.57–62 Raman band at high frequency A1g (679 cm−1) can be attributed to the characteristic vibration of oxygen atom inside the CoO6 octahedral unit,63 whereas F2g and Eg modes are due to the vibration of tetrahedral (CoO4) and octahedral sites.64 In sample (d), the width of the peaks is small indicating the formation of the perovskite. The major red shift in the Raman spectrum of catalyst (e) is because of factors like increase in particle size and crystallinity.65 The larger crystal size is also reflected in TPR, XRD and SEM Results. In the reduced samples the above characteristic peaks are conspicuously absent indicating the formation of metallic Co as a consequence of reduction.
 |
| | Fig. 5 Laser Raman of (A) calcined and (B) reduced catalysts; (a) La0.1Ce0.9CoO3 (b) La0.3Ce0.7CoO3 (c) La0.5Ce0.5CoO3(d) La0.7Ce0.3CoO3 (e) La0.9Ce0.1CoO3. | |
3.6 X-ray photoelectron spectroscopy
Co2p XPS spectra. Co2p XPS spectra are shown in Fig. 6. The core level peaks of the Co2p3/2 (B.E.: 779.2–780.2 eV) and Co2p1/2 (B.E.: 794.1–795 eV) observed for the calcined catalysts are quite identical with the B.Es reported for the CoO in equilibrium with Co3O4.66 The intensity of the satelite peaks positioned at ∼6 eV increased from (a) to (c) indicating that the proportion of the Co3O4 is more in these samples. Particularly, in sample (d) the intensity of the 791.1 eV peak increased and has become equal to the intensity of the 787.8 eV peak clearly indicating that cobalt existed as CoO along with Co3O4 in LaCoO3 perovskite.
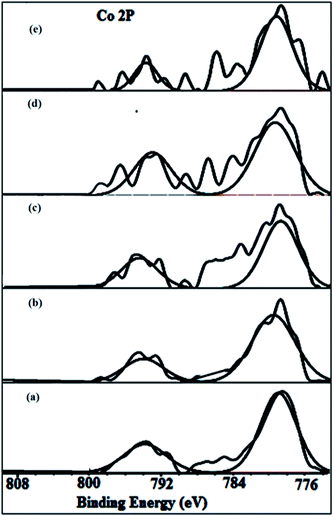 |
| | Fig. 6 Co 2p XPS spectra of calcined (a) La0.1Ce0.9CoO3 (b) La0.3Ce0.7CoO3 (c) La0.5Ce0.5CoO3 (d) La0.7Ce0.3CoO3 (e) La0.9Ce0.1CoO3. | |
O 1s XPS spectra. The spectra of O1s are presented in Fig. 7. Two B.Es are identified for oxygen; the first peak between 529.9–530.2 eV and the second between 531.1-531.9 eV. These peaks can be assigned to the O attached to the Co metal of CoO and the O present in water and surface adsorbed O–H group attached to the Co metal, respectively. From the O 1s XPS spectra it is observed that the B.E of the oxygen attached to Co metal increased with increasing the La2O3 content. The peak intensity of the lattice oxygen increased with La2O3 content in the catalysts.66
 |
| | Fig. 7 O 1s XPS spectra of calcined (a) La0.1Ce0.9CoO3 (b) La0.3Ce0.7CoO3 (c) La0.5Ce0.5CoO3 (d) La0.7Ce0.3CoO3 (e) La0.9Ce0.1CoO3. | |
3.7 UV-DRS
The UV-DRS spectra of the calcined and reduced samples are shown in Fig. 8(A) and (B), respectively. UV-DRS is an important technique to know the geometry of the metal centres existing in the material. The band found at 260 nm is purely due to the charge transfer of the oxygen to ligand in the metal oxide. The bands located at 380 and 660 nm resemble that of Co3O4 structure. The broad band obtained in the 540–750 nm region is a result of the combination of the Co2+ present in the tetrahedral (Td) and the Co3+ present in the octahedral position (Oh). The formation of the perovskite structure is evidenced with the blue shift of the 380 and 660 nm bands observed for the samples (d) and (e). This is because of the incorporation of Co in the perovskite geometry.67–69
 |
| | Fig. 8 UV-DRS of (A) calcined and (B) reduced catalysts; (a) La0.1Ce0.9CoO3 (b) La0.3Ce0.7CoO3 (c) La0.5Ce0.5CoO3 (d) La0.7Ce0.3CoO3 (e) La0.9Ce0.1CoO3. | |
The UV-Vis DRS spectra of the reduced samples do not reveal the characteristic bands of Co3+ or Co2+ belonging to the octahedral or tetrahedral sites. Only the charge transfer bands of the O → Ce (CeO2) and O → La (La2O3) vibrational bonds are present, as also reported in the literature.70,71 This information further supports the XRD and FT-IR results of the reduced samples.
3.8 Catalytic activity
Glycerol steam reforming is a metal-active reaction. Hence, metal dispersion plays a promising role. The values of dispersion of Co, as determined by the H2 pulse chemisorptions, are presented in Table 2. It can be observed that the active metal dispersion increased with increasing La content and the highest dispersion is achieved for the sample La0.7Ce0.3CoO3. Further increase of La decreased the metal dispersion. This could be due to the formation of the perovskite (LaCoO3) in the fresh sample and also the larger particle size.
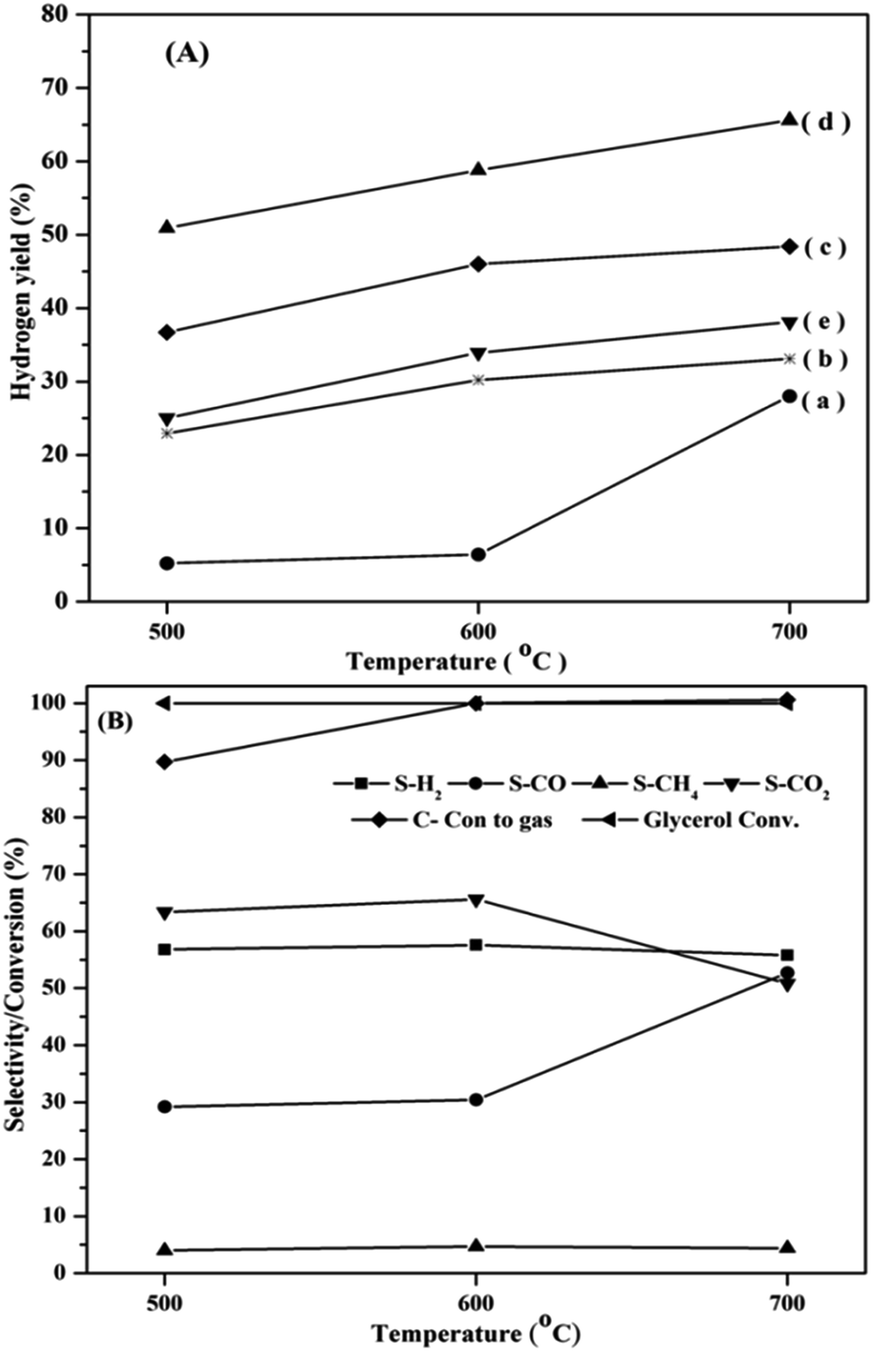
All the catalysts showed complete conversion of glycerol, due to high reaction temperature. However, the hydrogen yield varied with temperature, as shown in Fig. 9(A). The hydrogen yield increased with increasing temperature because of the endothermic nature of the reaction.
 |
| | Fig. 9 (A) & (B) activity results of; (a) La0.1Ce0.9CoO3 (b) La0.3Ce0.7CoO3 (c) La0.5Ce0.5CoO3 (d) La0.7Ce0.3CoO3 (e) La0.9Ce0.1CoO3. | |
The progress of steam reforming depends on the reducibility of the metal oxide.72 Among, all the catalysts studied La0.7Ce0.3CoO3 exhibited the highest hydrogen yield (68%) at 700 °C. This is due the presence of easily reducible Co species with suitable crystal dimensions. When the fresh samples are reduced at higher temperatures, the perovskite structure collapses leading to the formation of well dispersed Co species,73–75 as evidenced by the XRD patterns of the reduced catalysts.
The distribution of gases in the overall reaction products is depicted in Fig. 9(B). The entire carbon in glycerol is converted to carbon products beyond 600 °C, at which the CO2 formation started decreasing with simultaneous increase in CO composition. The selectivity towards hydrogen almost remained constant. An interesting feature of the catalyst is that the amount of methane formed is very low.
In order to verify the effect of dispersion on the activity, hydrogen chemisorption was carried out on these catalysts. The Co dispersion and particle size are presented in Table 2. It is interesting to observe that the major portion of total surface area of the reduced catalyst (approx. 80%) is due to the contribution of small Co particles. That is the reason why the BET area of the reduced catalysts follows similar trend as that of the fresh catalysts. The H2 uptake which is used to calculate the active metal area follows similar trend (Fig. 10(A)). Fig. 10(B) shows a nearly parallel behavior existing between the metal surface area and the hydrogen yield, when drawn against the La content in the catalysts. It can be observed from the Table 2 that the dispersion is the highest in the case of La0.7Ce0.3CoO3 catalyst.
 |
| | Fig. 10 (A) The variation of H2 uptake, (B) metal surface area and hydrogen yield with La content. | |
Turn over frequency (TOF), is a measure of rate of H2 production per active cobalt site per second. In the present study.
H2-chemisorption technique was used to find out the number of surface cobalt atoms. TOF is found to be sensitive to the particle size. Literature also reports that the activity of carbon nano fibre supported Co catalyst in the FT synthesis depends on the Co particle size and the TOF with respect to hydrogen consumption increases with increase in Co particle size and reaches a maximum when Co particle size is 6 nm.76 In the present investigation also, a particle size of 4–5 nm seems to be the optimum value for getting high TOF. This behaviour is displayed in Fig. 11, drawn to explain the variation of TOF with Co metal area. The turnover frequency increased steadily with metal surface area (upto 40.7 m2 g−1), beyond which a sudden jump is noticed. The rate of hydrogen production reached its maximum (68% yield) as the particle size corresponding to the metal area of 45.2 m2 g−1 is approached. The highest metal area is achieved for the catalyst La0.7Ce0.3CoO3. Table 2 also discloses the extent of coke formation, analyzed after the reaction. It is also found to be the lowest for this catalyst.
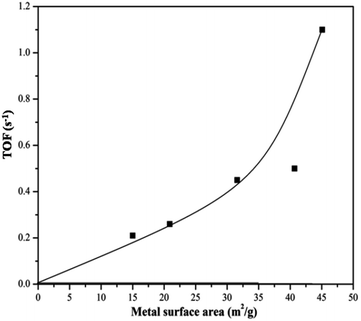 |
| | Fig. 11 Variation of TOF with metal surface area. | |
Glycerol conversion and hydrogen yield depend upon the nature of the catalyst, the steam/glycerol (S/C) ratio and reaction temperature. Even though, a direct comparison is not possible, Table 3 displays the recent literature reports on hydrogen generation from glycerol using different catalysts along with the optimum catalyst identified in this work. It can be observed that La0.7Ce0.3CoO3 compares well or even better in some cases.
Table 3 Activity comparison with literature reported values
| S. no |
Catalysts |
Reaction conditions |
Glycerol conversion |
Hydrogen |
References |
| 1 |
NiO (24.1%)–MgO (26.1%) Al2O3 (49.8%) |
650 °C, 1 atm 1.0 g, 10 wt% glycerol |
88.0% at 650 °C |
Selectivity 78.5% |
77 |
| 2 |
La0.5Ca0.5NiO3 |
550 °C, 1 atm S/C = 3.0 |
100% at 550 °C |
Yield 82.2% |
78 |
| 3 |
Ni 29.2 wt%–Cu 31.1 wt%–Al 39.7 wt% |
600 °C, 1 atm, 1.0g S/C = 3.0 |
71.9% at 600 °C |
Selectivity 78.6% |
79 |
| 4 |
La0.5Ce0.5NiO3 |
700 °C,1 atm, 1.0 g 30 wt% |
88% at 700 °C |
Selectivity 70% |
80 |
| 5 |
La0.7Ce0.3CoO3 |
700 °C, 1 atm, 1.0 g 30 wt% glycerol, 0.08 mL min−1 |
100% at 700 °C |
Yield 68% |
Present study |
4. Conclusions
The Co catalysts derived from La–Ce–Co mixed oxides are active for the glycerol steam reforming producing hydrogen-rich gas products. Characterization of reduced catalysts reveals the structure collapse and dispersion of smaller Co metallic particles on the mixed oxide (La2O3–CeO2) support. It is possible to tune the dispersion of Co metal (obtained after reduction of the fresh catalysts), and thus the reforming activity and coke formation by using different Co-containing mixed oxides as starting materials. Lanthana plays a vital role in the enhancement of metal active surface area as well as the dispersion. La0.7Ce0.3CoO3 is found to be the best initial composition for arriving at the maximum hydrogen yield. The active metal area with major share in total area of the reduced samples correlates well with the TOF. It can be concluded that the Co metal dispersion is proportional to hydrogen yield and minimizes coke formation on the catalyst surface.
Acknowledgements
The authors thank Council of scientific industrial research & Department of Science and Technology (DST), New Delhi.
References
- B. Zhang, X. Tang, Y. Li, Y. Xu and W. Shen, Int. J. Hydrogen Energy, 2007, 32, 2367 CrossRef CAS PubMed.
- P. R. d. l. Piscina and N. Homs, Chem. Soc. Rev., 2008, 37, 2459 RSC.
- C. H. Zhou, J. N. Beltramini, Y. X. Fan and G. Q. Lu, Chem. Soc. Rev., 2008, 37, 527 RSC.
- J. W. Shabaker, G. W. Huber and J. A. Dumesic, J. Catal., 2004, 222, 180 CrossRef CAS PubMed.
- J. Fermoso, L. He and D. Chen, Int. J. Hydrogen Energy, 2012, 37, 14047 CrossRef CAS PubMed.
- R. D. Cortright, R. R. Davda and J. A. Dumesic, Nature, 2002, 418, 964 CrossRef CAS PubMed.
- G. W. Huber, J. W. Shabaker and J. A. Dumesic, Science, 2003, 300, 2075 CrossRef CAS PubMed.
- R. R. Soares, D. A. Simonetti and J. A. Dumesic, Angew. Chem., Int. Ed., 2006, 45, 3982 CrossRef CAS PubMed.
- A. O. Menezes, M. T. Rodrigues, A. Zimmaro, L. E. P. Borges and M. A. Fraga, Renewable Energy, 2011, 36, 595 CrossRef CAS PubMed.
- D. A. Simonetti, E. L. Kunkes and J. A. Dumesic, J. Catal., 2007, 247, 298 CrossRef CAS PubMed.
- S. Adhikari, S. Fernando and A. Haryanto, Energy Fuels, 2007, 21, 2306 CrossRef CAS.
- G. R. Moradi and M. Rahmanzadeh, Catal. Commun., 2012, 26, 169 CrossRef CAS PubMed.
- T. Borowieck, Appl. Catal., 1984, 10, 273 CrossRef.
- S. Adhikari, S. Fernando and A. Haryanto, Catal. Today, 2007, 129, 355 CrossRef CAS PubMed.
- B. Zhang, X. Tang, Y. Li, Y. Xu and W. Shen, Int. J. Hydrogen Energy, 2007, 32, 2367 CrossRef CAS PubMed.
- M. Slinn, K. Kendall, C. Mallon and J. Andrews, Bioresour. Technol., 2008, 99, 5851 CrossRef CAS PubMed.
- S. Adhikari, S. D. Fernando and A. Haryanto, Renewable Energy, 2008, 33, 1097 CrossRef CAS PubMed.
- A. Iriondo, V. L. Barrio, J. F. Cambra, P. L. Arias, M. B. Güemez, R. M. Navarro, M. C. Sánchez-Sánchez and J. L. G. Fierro, Top. Catal., 2008, 49, 46 CrossRef CAS.
- B. Dou, C. Wang, H. Chen, Y. Song and B. Xie, Int. J. Hydrogen Energy, 2013, 38, 11902 CrossRef CAS PubMed.
- C. Wang, B. Dou, H. Chen, Y. Song, Y. Xu, X. Du, L. Zhang, T. Luo and C. Tan, Int. J. Hydrogen Energy, 2013, 38, 3562 CrossRef CAS PubMed.
- B. Dou, Y. Song, C. Wang, H. Chen and Y. Xu, Renewable Sustainable Energy Rev., 2014, 30, 950 CrossRef CAS PubMed.
- P. Y. Hwa, K. J. Yeon, M. D. Ju, P. N. Cook and K. Y. Chul, Res. Chem. Intermed., 2015 DOI:10.1007/s11164-015-2020-7.
- G. Wu, C. Zhang, S. Li, Z. Han, T. Wang, X. Ma and J. Gong, ACS Sustainable Chem. Eng., 2013, 1, 1052 CrossRef CAS.
- U. Ahmed and T. S. John Irvine, Energy Fuels, 2013, 58, 187 Search PubMed.
- C. Wang, B. Dou, H. Chen, Y. Song, Y. Xu, X. Du, T. Luo and C. Tan, Chem. Eng. J., 2013, 220, 133 CrossRef CAS PubMed.
- Ji, W.-rong, Zeng, Yu-long, Tan, Ye-bin, Ying, Hui-juan, Zhang, Ying From Zhejiang Gongye Daxue Xuebao, 2013, 41, 6.
- Z.-Y. Huang, C.-H. Xu, C.-Q. Liu, H.-W. Xiao, J. Chen, Y.-X. Zhang and Ya-C. Lei, Korean J. Chem. Eng., 2013, 30, 587 CrossRef CAS.
- C. A. Franchini, W. Aranzaez, A. M. D. Farias, G. Pecchi and M. A. Fraga, Appl. Catal., B, 2014, 147, 193 CrossRef CAS PubMed.
- G. Wu, S. Li, C. Zhang, T. Wang and J. Gong, Appl. Catal., B, 2014, 144, 277 CrossRef CAS PubMed.
- V. V. Thyssen, T. A. Maia and E. M. Assaf, Fuel, 2013, 105, 358 CrossRef CAS PubMed.
- S. Kitamura, T. Su-enaga, N. Ikenaga, T. Miyake and T. Suzuki., Catal. Lett., 2011, 141, 895 CrossRef CAS PubMed.
- A. Iriondo, M. B. Güemez, V. L. Barrio, J. F. Cambra, P. L. Arias, M. C. Sánchez-Sánchez, R. M. Navarro and J. L. G. Fierro, Stud. Surf. Sci. Catal., 2010, 175, 449 CAS.
- S. Ramesh, E. H. Yang, J. S. Jung and D. J. Moon, Int. J. Hydrogen Energy DOI:10.1016/j.ijhydene.2015.02.013 , article in press.
- Y. Cui, V. Galvita, L. Rihko-Struckmann, H. Lorenz and K. Sundmacher, Appl. Catal., B, 2009, 90, 29 CrossRef CAS PubMed.
- M. R. Goldwasser, M. E. Rivas, E. Pietri, M. J. Pérez-Zurita, M. L. Cubeiro, L. Gingembre, L. Leclercq and G. Leclercq, Appl. Catal., 2003, 225, 45 CrossRef.
- E. Pietri, A. Barrios, O. González, M. Goldwasser, M. Pérez-Zurita, M. Cubeiro, J. Goldwasser, L. Leclercq, G. Leclercq and L. Gingembre, Stud. Surf. Sci. Catal., 2001, 136, 381 CAS.
- S. M. de Lima, I. O. da Cruz, G. Jacobs, B. H. Davis, L. V. Mattos and F. B. Noronha, J. Catal., 2008, 257, 356 CrossRef CAS PubMed.
- P. Biswas and D. Kunzru, Chem. Eng. J., 2008, 136, 41 CrossRef CAS PubMed.
- D. Srinivas, C. V. V. Satyanarayana, H. S. Potdar and P. Ratnasamy, Appl. Catal., A, 2003, 246, 323 CrossRef CAS.
- C. Diagne, H. Idriss and A. Kiennemann, Catal. Commun., 2002, 12, 565 CrossRef.
- N. Srisiriwata, S. Therdthianwonga and A. Therdthianwong, Int. J. Hydrogen Energy, 2009, 34, 2224 CrossRef PubMed.
- B. Valle, A. Remiro, A. T. Aguayo, J. Bilbao and A. G. Gayubo, Int. J. Hydrogen Energy, 2013, 38, 1307 CrossRef CAS PubMed.
- C. A. Franchinia, W. Aranzaezb, A. M. D. de Fariasa, G. Pecchi and M. A. Fragaa, Appl. Catal., B, 2014, 147, 193 CrossRef PubMed.
- B. Seyfi, M. Baghalhaa and H. Kazemian, Chem. Eng. J., 2009, 148, 306 CrossRef CAS PubMed.
- P. Jana, V. A. P. OShea, J. M. Coronado and D. P. Serrano, Appl. Catal., A, 2013, 467, 371 CrossRef CAS PubMed.
- V. V. Matveev, D. A. Baranov, G. Y. Yurkov, N. G. Akatiev, I. P. Dotsenko and S. P. Gubin, Chem. Phys. Lett., 2006, 422, 402 CrossRef CAS PubMed.
- L. Huang, M. Bassir and S. Kaliaguine, Mater. Chem. Phys., 2007, 101, 259 CrossRef CAS PubMed.
- S. S. Y. Lin, D. H. Kim and S. Y. Ha, Appl. Catal., A, 2009, 355, 69 CrossRef CAS PubMed.
- N. E. Machin, C. Karakaya and A. Celepci, Energy Fuels, 2008, 22, 2166 CrossRef CAS.
- Z. Orel, Internet J. Vib. Spectrosc., 1999, 3, 4 Search PubMed , http://www.ijvs.com.
- A. B. P. Lever, E. Mantovani and B. S. Ramaswamy, Can. J. Chem., 1971, 49, 1957 CrossRef CAS.
- B. Yan and H. Zhu, J. Nanopart. Res., 2008, 10, 1279 CrossRef CAS.
- B. Klingenberg and M. A. Vannice, Chem. Mater, 1996, 8, 2755 CrossRef CAS.
- R. N. S. H. Magalhães, F. S. Toniolo, V. T. Silva and M. Schmal, Appl. Catal., A, 2010, 388, 216 CrossRef PubMed.
- L. Huang, M. Bassir and S. Kaliaguine, Mater. Chem. Phys., 2007, 101, 259 CrossRef CAS PubMed.
- J. A. Villoria, M. C. Alvarez-Galvan, S. M. Al-Zahrani, P. Palmisano, S. Specchia, V. Specchia, J. L. G. Fierro and R. M. Navarro, Appl. Catal., B, 2011, 105, 276 CrossRef CAS PubMed.
- L. Xi, Z. Peng, W. Fan, K. Guo, J. Gu, M. Zhao and J. Meng, Mater. Chem. Phys., 1996, 46, 50 CrossRef.
- N. Orlovskaya, D. Steinmetz, S. Yarmolenko, D. Pai, J. Sankar and J. Goodenough, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 014122 CrossRef.
- A. Ishikawa, J. Nohara and S. Sugai, Phys. Rev. Lett., 2000, 93, 1193 Search PubMed.
- M. Popa, J. Frantti and M. Kakihana, Solid State Ionics, 2002, 154, 135 CrossRef.
- W. W. -Ran, X. D. Peng, S. W. Hui, D. Z. Hui, X. Y. Feng and S. G. Xin, Chin. Phys. Lett., 2005, 22, 2400 CrossRef.
- D. P. Kozlenko, N. O. Golosova, Z. Jirák, L. S. Dubrovinsky, B. N. Savenko, M. G. Tucker, Y. Le Godec and V. P. Glazkov, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 064422 CrossRef.
- M. Christy, M. R. Jisha, A. R. Kim, K. S. Nahm, D. J. Yoo, E. K. Suh, T. S. D. Kumari, T. P. Kumar and A. M. Stephan, Bull. Korean Chem. Soc., 2011, 32, 1204 CrossRef CAS.
- C. F. Windisch, G. J. Exarhos, K. F. Ferris, M. H. Engelhard and D. C. Stewart, Thin Solid Films, 2001, 398, 45 CrossRef.
- I. Lopes, N. El Hassan, H. Guerba, G. Wallez and A. Davidson, Chem. Mater., 2006, 18, 5826 CrossRef CAS.
- C. C. Li, X. M. Yin, T. H. Wang and H. C. Zeng, Chem. Mater., 2009, 21, 4984 CrossRef CAS.
- K. Kaviyarasu, A. Raja and P. A. Devarajan, Spectrochim. Acta, Part A, 2013, 114, 586 CrossRef CAS PubMed.
- W. Young Jung and S.-S. Hong, J. Ind. Eng. Chem., 2013, 19, 157 CrossRef PubMed.
- J. Yan, M. C. Kung, W. M. H. Sachtler and H. H. Kung, J. Catal., 1997, 172, 178 CrossRef CAS.
- M. Guo, J. Lu, Y. Wu, Y. Wang and M. Luo, Langmuir, 2011, 27, 3872 CrossRef CAS PubMed.
- S. Satish, D. K. Pradhan, G. Hota and B. G. Mishra, Chem. Eng. J., 2012, 193, 1 Search PubMed.
- L. P. R. Profeti, E. A. Ticianelli and E. M. Assaf, Int. J. Hydrogen Energy, 2009, 34, 5049–5060 CrossRef CAS PubMed.
- G. Valderrama, M. R. Goldwasser, C. U. d. Navarro, J. M. Tatibouët, J. Barrault, C. Batiot-Dupeyrat and F. Martínez, Catal. Today, 2005, 107–108, 785–791 CrossRef CAS PubMed.
- G. S. Gallego, F. Mondragón, J. Barrault, J.-M. Tatibouët and C. Batiot-Dupeyrat, Appl. Catal., A, 2006, 311, 164–171 CrossRef CAS PubMed.
- R. M. Navarro, M. C. Alvarez-Galvan, J. A. Villoria, I. D. González-Jiménez, F. Rosa and J. L. G. Fierro, Appl. Catal., B, 2007, 73, 247–258 CrossRef CAS PubMed.
- G. L. Bezemer, J. H. Bitter, H. P. C. E. Kuipers, H. Oosterbeek, J. E. Holewijn, X. Xu, F. Kapteijn, A. J. V. Dillen and K. P. de Jong, J. Am. Chem. Soc., 2006, 128, 3956 CrossRef CAS PubMed.
- C. Wang, B. Dou, H. Chen, Y. Song, Y. Xu, X. Du, T. Luo and C. Tan, Chem. Eng. J., 2013, 220, 133 CrossRef CAS PubMed.
- G. Wua, S. Lia, C. Zhanga, T. Wanga and J. Gong, Appl. Catal., B, 2014, 144, 277 CrossRef PubMed.
- B. Dou, C. Wang, Y. Song, H. Chen and Y. Xu, Energy Convers. Manage., 2014, 78, 253 CrossRef CAS PubMed.
- C. A. Franchini, W. Aranzaez, A. M. D. de Fariasa, G. Pecchi and M. A. Fraga, Appl. Catal., B, 2014, 147, 193 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 




 c
c  Nm = the number of adsorbed gas molecules, S = adsorption stoichiometry, Am = the cross-sectional area occupied by each active metal surface atom, M and L are the molecular weight and percent loading of the supported metal, f is a particle shape correction factor (=6 for spherical particles). Z = density of the supported metal.
Nm = the number of adsorbed gas molecules, S = adsorption stoichiometry, Am = the cross-sectional area occupied by each active metal surface atom, M and L are the molecular weight and percent loading of the supported metal, f is a particle shape correction factor (=6 for spherical particles). Z = density of the supported metal.