
Firmly bonded graphene–silicon nanocomposites as high-performance anode materials for lithium-ion batteries
Abstract
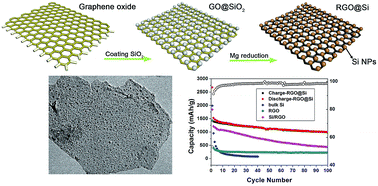
We demonstrate the synthesis of firmly bonded reduced graphene oxide (RGO)@Si nanocomposites via the magnesiothermic reduction of graphene oxide (GO)@SiO2 nanocomposites. The uniform deposition of SiO2 layer on the GO nanosheets is achieved via controllable TEOS hydrolysis, which is a prerequisite for the synthesis of uniform RGO@Si nanocomposites. When used as an anode material for lithium-ion batteries, the as-synthesized RGO@Si nanocomposites show high reversible capacity and good cycling performance, which is better than bare Si nanoparticles and Si/RGO nanocomposites synthesized from physical blending Si nanoparticles and RGO nanosheets. It is believed that the improved electrochemical performance can be attributed to the novel uniform nanostructure and the introduction of RGO multilayers that can mitigate the volume expansion/contraction and enhance the electronic conductivity of Si anode materials.
 Please wait while we load your content...
Please wait while we load your content...

