
I2/Cu-mediated self-sorting domino reaction of aryl β-ketoesters into symmetrical 2-carboalkoxy-1,4-enediones: application to synthesis of pyrazine, β-carboline and quinoxalines†
Gandhesiri Satish and
Andivelu Ilangovan*
School of Chemistry, Bharathidasan University, Tiruchirappalli – 620 024, Tamil Nadu, India. E-mail: ilangovanbdu@yahoo.com
First published on 18th May 2015
Abstract
A self-sorting domino reaction of aryl β-ketoesters into symmetrical 1,4-enediones is reported by an I2/Cu system. The reaction proceeds through tandem iodination, self-dimerization and Krapcho dealkoxycarbonylation in one pot under open air condition. Further, 1,4-enediones were successfully employed for the synthesis of bioactive pyrazine, β-carboline and quinoxalines via aza-Michael addition, intramolecular cyclization and C–C bond cleavage of 1,3-dicarbonyl unit under mild reaction condition.
Introduction
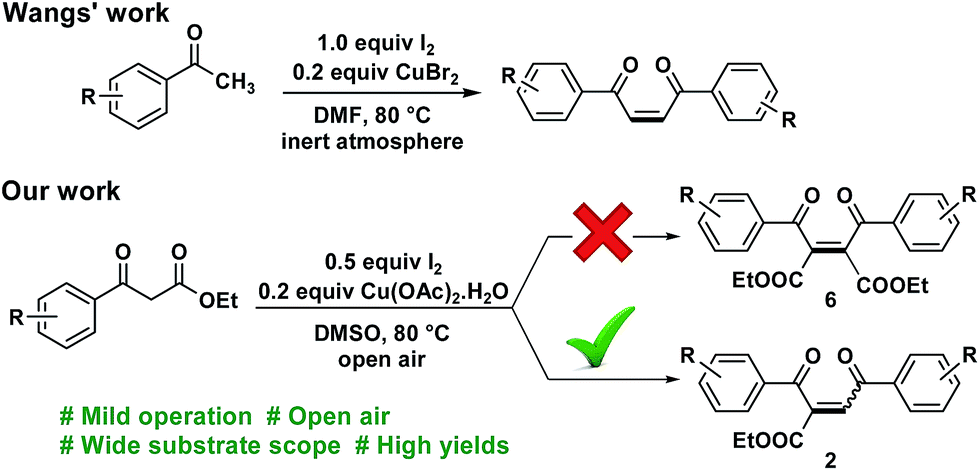
Development of simplified strategies for the synthesis of complex molecules is always an exciting and challenging task. In the past few decades a significant development of domino reactions was witnessed, mainly due to its influent advantages such as, high atom economy, reduction of labour, easy resource management and production of complex molecules in one pot.1 Although, different type of domino approaches namely intramolecular, intermolecular, multicomponent, and focusing were well established, studies on self-sorting domino (SSD) reactions are only limited.2 This is because, in a SSD reaction one should manage to control the reactivity of in situ generated intermediates from a single starting material to get the desired product in high yield. As this helps effective utilization of a single starting material, SSD reaction could be considered as one of the most efficient domino reactions.1,4-Enedione is a privileged structural motif found among scores of bio-active compounds, and marine natural products.3 It has been extensively used as an admirable starting material for the synthesis of enamine diones, oxygen and nitrogen heterocycles, etc. by conjugate addition of carbon, phosphorus and nitrogen nucleophiles (Fig. 1).4 Traditional approaches followed for the synthesis of 1,4-enediones are ring opening of furan or thiophene derivatives,5 decomposition of diazo carbonyl compounds6 and oxidation of enone unit.7 Recently, Kirsch et al. reported the construction of Z-enediones in moderate yield by oxidative rearrangement of 2-alkynyl alcohols.8 Similarly, Wang et al. described the complete E-selective synthesis of 1,4-enediones from aromatic methyl ketones via a tandem pathway (Scheme 1).9 Notably, Wu's research group reported the synthesis of E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z mixture of unsymmetrical 1,4-enediones by reaction of 1,3-dicarbonyl compounds with acetophenones/styrenes/secondary alcohols/α-halo acetophenones via Kornblum oxidation followed by Knoevenagel condensation.10 While these investigations are impressive, the need of multiple substrates and stoichiometric amount of reagents necessitates the development of a simple and sustainable protocol for the construction of 1,4-enediones.
:Z mixture of unsymmetrical 1,4-enediones by reaction of 1,3-dicarbonyl compounds with acetophenones/styrenes/secondary alcohols/α-halo acetophenones via Kornblum oxidation followed by Knoevenagel condensation.10 While these investigations are impressive, the need of multiple substrates and stoichiometric amount of reagents necessitates the development of a simple and sustainable protocol for the construction of 1,4-enediones.
 | ||
| Fig. 1 Synthetic utility of 1,4-enediones. | ||
 | ||
| Scheme 1 Synthetic strategies of 1,4-enediones. | ||
On the other hand, the cleavage of unstrained and unactivated carbon–carbon σ bonds under mild reaction conditions is a challenging task and such reactions are only few.11 Under the methods reported so far, the C–C bond cleavage is achieved by using activating group and driven by relieving steric strain12 or employing high temperature/pressure.13 Very recently, study on C–C bond cleavage of 1,3-dicarbonyl unit has gained much importance14 and such cleavage was reported by Kotora,14a Zhai,14 Li14c,d and Wu14e separately by employing various catalytic systems.
Based on our previous experience of C–H activation of 2′-aminoacetophenones using Cu(OAc)2·H2O15a and I2,15b we envisaged that aryl β-ketoester in the presence of I2/Cu system would undergo self dimerization to give diethyl 2,3-dibenzoylmaleate (10). However, to our surprise, the reaction furnished 2-carboalkoxy-1,4-enediones (2) via self dimerization followed by Krapcho dealkoxycarbonylation in one pot.16 To the best of our knowledge, a self-sorting domino reaction of aryl β-ketoester as well as Krapcho dealkoxycarbonylation in the presence of I2/Cu system was observed for the first time. Considering the rare occurrence of SSD reaction and synthetic importance of 1,4-enedione, herein we report a novel SSD reaction of aryl β-ketoesters into symmetrical 1,4-enediones. Further, 1,4-enediones were underwent C–C bond cleavage of 1,3-dicarbonyl unit to give rise biologically important pyrazine, β-carboline and quinoxalines in high yields.
Results and discussion
Our initial study began with the reaction of ethyl benzoylacetate (1a) with I2 (1.0 equiv.) and Cu(OAc)2·H2O (1.0 equiv.) in DMSO at room temperature (Table 1, entry 1). Since no reaction took place at r.t., the reaction temperature was raised to 80 °C and we were heartened to know that self dimerization followed by dealkoxycarbonylation took place to give 1,4-enedione 2a in 68% isolated yield (entry 2). There was no appreciable change in the yield when quantity of I2 and Cu(OAc)2·H2O was decreased to 0.5 equiv. (entry 3). Interestingly, the yield was improved to 78% by lowering the quantity of Cu(OAc)2·H2O to 0.2 equiv. (entry 4). Increasing or decreasing the temperature from 80 °C, only led to decrease in yield (entries 5 and 6). In the absence of either I2 or Cu(OAc)2·H2O a mixture of products was obtained which indicates that both are essential for this reaction (entries 7 and 8). Screening various copper salts revealed that Cu(OAc)2·H2O is the superior for this reaction (entries 9–14). Among different solvents, DMSO furnished highest yield (entries 15–18). As a result of the screening study, I2 (0.5 equiv.), and Cu(OAc)2·H2O (0.2 equiv.) in DMSO at 80 °C was found to be the optimum condition for further study.
|
||||
|---|---|---|---|---|
| Entry | Iodine (equiv.) | Copper salt (equiv.) | Solvent | Temp [°C]/time [h]/yieldb [%] |
| a Unless otherwise stated, all the reactions were performed using 1a (1.0 mmol) in solvent (3.0 mL).b Isolated yield.c No reaction.d Not determined. | ||||
| 1c | I2 (1.0) | Cu(OAc)2·H2O (1.0) | DMSO | r.t./20/n.r. |
| 2 | I2 (1.0) | Cu(OAc)2·H2O (1.0) | DMSO | 80/8/68 |
| 3 | I2 (0.5) | Cu(OAc)2·H2O (0.5) | DMSO | 80/10/70 |
| 4 | I2 (0.5) | Cu(OAc)2·H2O (0.2) | DMSO | 80/12/78 |
| 5 | I2 (0.5) | Cu(OAc)2·H2O (0.2) | DMSO | 100/7/70 |
| 6 | I2 (0.5) | Cu(OAc)2·H2O (0.2) | DMSO | 60/16/68 |
| 7 | I2 (0.5) | — | DMSO | 80/15/43 |
| 8d | — | Cu(OAc)2·H2O (0.2) | DMSO | 80/12/n.d. |
| 9 | I2 (0.5) | CuCl2 (0.2) | DMSO | 80/15/42 |
| 10 | I2 (0.5) | CuBr2 (0.2) | DMSO | 80/12/57 |
| 11 | I2 (0.5) | Cu(NO3)2 (0.2) | DMSO | 80/18/38 |
| 12 | I2 (0.5) | Cu(SO4)2 (0.2) | DMSO | 80/20/trace |
| 13 | I2 (0.5) | CuI (0.2) | DMSO | 80/12/48 |
| 14 | I2 (0.5) | CuBr (0.2) | DMSO | 80/12/36 |
| 15 | I2 (1.0) | Cu(OAc)2·H2O (0.2) | DMF | 80/24/61 |
| 16 | I2 (1.0) | Cu(OAc)2·H2O (0.2) | Toluene | 80/24/48 |
| 17 | I2 (1.0) | Cu(OAc)2·H2O (0.2) | CH3OH | 80/24/42 |
| 18d | I2 (1.0) | Cu(OAc)2·H2O (0.2) | CH3CN | 80/24/n.d. |

Under the optimal reaction condition, the substrate scope of various aryl β-ketoesters was investigated (Table 2). As shown, compound 1b with phenyl substituent underwent slow reaction to give 1,4-enedione 2b in 71% yield. Substrates 1c–i bearing electron donating and halo substituent's provided 1,4-enediones 2c–i in high yield. The electron withdrawing NO2 group in 1j was well tolerated the present reaction condition and offered corresponding enedione 2j in 69% yield. The β-ketoesters 1k–m with substituent in sterically and electronically disadvantaged ortho and meta position afforded expected 1,4-enediones 2k–m in moderate to good yield. Dimethyl substituted β-ketoester 1n gave the desired product 2n in 76% yield. Aryl β-ketoester 1o containing heterocyclic thiophene motif well tolerated the optimal condition and corresponding 1,4-enedione 2o was obtained in 78% yield. Similarly, ethyl naphthoylacetate (1p) gave desired enedione 2p in 75% yield. Further, similar to ethyl ester 1e, the corresponding methyl ester 1q also provided the expected product 2q in almost same yield. However, the aliphatic β-ketoester, ethyl acetoacetate (1r) failed to deliver the desired product 2r. All the substrates 1a–q were provided the corresponding 1,4-enediones 2a–q, as E/Z mixture, and the thermodynamically stable E-isomers were the major products. This was confirmed from the 1H-NMR spectra.

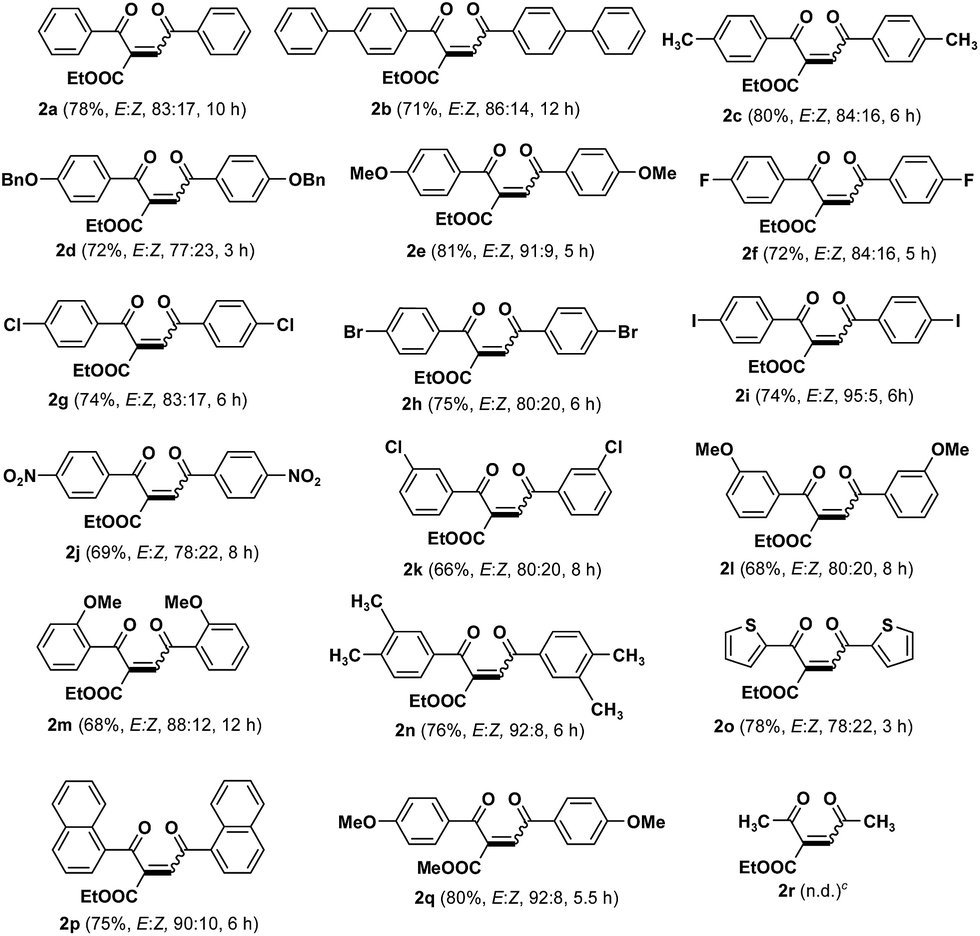
The present method is applicable to a wide variety of aryl β-ketoesters containing different electron donating groups such as 4-OMe, 4-OBn, 4-CH3, 3,4-dimethyl, sterically hindered 2-OMe, biphenyl, naphthyl, heterocyclic (thiophene), inductively electron withdrawing halogens and electron withdrawing 4-NO2 groups.
Further, to demonstrate the synthetic utility of obtained 1,4-enediones (2), we have examined an iodine-mediated intermolecular nucleophilic addition of 1,4-enedione 2a with 1,2-diamino benzene (3a) (Table 3). To our delight, the reaction furnished quinoxaline 4a by loss of a 1,3-dicarbonyl unit at room temperature. Since quinoxaline is an important structural motif in many drugs,17 organic semiconductors,18 electroluminescent materials,19 and synthetic dyes,20 we investigated the reaction to improve the yield by employing other iodine sources, such as NIS and TBAI. However none of them provided the desired product. DMSO was the choice of solvent to get best yield of quinoxaline 4a. Under suitable condition, 1,4-enedione bearing electron rich and halo groups provided quinoxalines 4b–e in 80–84% yields. Steric effect of 1,4-enediones did not influence the rate of reaction and corresponding quinoxalines 4f and 4g were obtained in 82% and 77% yield respectively. Thiophene motif containing 1,4-enedione 2m was well tolerated and delivered the corresponding quinoxaline 4h in 82% yield. The reaction of 3-methyl-1,2-diamino benzene (3b) with 2a and 2e afforded the corresponding quinoxalines 4i (4ia + 4ib) and 4j (4ja + 4jb) as regio-isomeric mixtures.


Next, we examined the reaction feasibility of 1,4-enedione 2j with 1,2-diamino ethane (5) (Scheme 2). Delightfully, the expected pyrazine 6 was obtained by loss of a 1,3-dicarbonyl unit followed by oxidation in 72% isolated yield. Importantly, the present method can also be applied in the total synthesis of natural product, eudistomin Y1. As shown in Scheme 2, the reaction of 1,4-enedione 2e with tryptamine (6) provided β-carboline 8 in good yield by losing 1,3-dicarbonyl unit. Further, β-carboline 8 was transformed to eudistomin Y1 according to the reported procedure.21
 | ||
| Scheme 2 Synthetic application of 1,4-enediones for pyrazine and β-carboline synthesis. | ||
To gain insight into the mechanism for the formation of 1,4-enedione (2), some control experiments were carried out (Scheme 3). Initially the α-iodo-β-ketoester 9 was prepared by iodination of ethylbenzoyl acetate (1a, Scheme 3, eqn (1)), further which on treatment with I2 (0.5 equiv.) and Cu(OAc)2·H2O (0.2 equiv.) provided diethyl 2,3-dibenzoylmaleate (10) in 84% isolated yield (Scheme 3, eqn (2)). When the reaction of 1a was stopped prematurely after 2 h, compound 10 was obtained in 80% yield (Scheme 3, eqn (3)). However, in the presence of TEMPO, the formation of 10 was drastically decreased, indicates that the reaction takes place through a radical pathway (Scheme 3, eqn (4)). In the absence of either I2 or Cu(OAc)2·H2O a mixture of products were obtained (Scheme 3, eqn (5) and (6)). When compound 10 was treated with 0.5 equiv. of I2 in d-DMSO gave the desired product 2a only in 32% yield (Scheme 3, eqn (7)). Interestingly, when same reaction was carried out in d-DMSO and H2O mixture, it gave 2a in 84% yield by dealkoxycarbonylation (Scheme 3, eqn (8)).16 These two reactions clearly indicate that, the carbanion is protonized by H2O and not DMSO.
 | ||
| Scheme 3 Controlled experiments to study reaction mechanism. | ||
On the basis of above information and the literature knowledge,9 a plausible mechanism is proposed (Scheme 4). Initially, ethylbenzoyl acetate (1a) undergoes α-iodination to give 9, which further may give radical A.22 Subsequently, radical A may undergo self dimerization to give 2,3-dibenzoyl-succinic acid diethyl ester B.23 Again B on α-iodination followed by copper insertion is expected to give radical C, which further may undergo easy deprotonation to form diethyl 2,3-dibenzoylmaleates 10.
 | ||
| Scheme 4 Proposed mechanism. | ||
Finally, the iodide ion, which is generated during the course of reaction, is expected to react with 10 to afford the desired product 2a by losing EtI and CO2 via Krapcho dealkoxycarbonylation.16 When 2a was treated with 1,2-diamino benzene (3a) in the presence of I2, it undergoes a sequential aza-Michael addition (D) followed by intramolecular cyclization to form the intermediate E. Finally, E provides quinoxaline 4a by losing ethylbenzoyl acetate 1a.
Conclusion
In conclusion, we have developed a practical and efficient self-sorting domino reaction of commercially available aryl β-ketoesters into symmetrical 2-carboalkoxy-1,4-enediones under open air condition, using simple catalytic system. The method is mild and flexible for the synthesis of a variety of symmetrical 1,4-enediones. The reaction proceeds through tandem iodination, self dimerization and Krapcho dealkoxycarbonylation in one pot. A suitable reaction mechanism is proposed by means of control experiments. Further, the synthetic utility of 1,4-enedione as an intermediate is successfully demonstrated by its conversion into a variety of biologically active β-carboline, pyrazine and quinoxalines via aza-Michael addition, intramolecular cyclization and C–C bond cleavage in a single reactor. Further mechanistic studies of this reaction are under way.Experimental
General information
All the reagents were purchased commercially and used as received. 1H NMR and 13C NMR were recorded with Bruker 400 MHz. 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded in CDCl3 with tetramethylsilane as the internal standard. Multiplicities are reported using the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad resonance. All the NMR spectra were acquired at ambient temperature. Analytical thin-layer chromatography (TLC) was performed using Silica Gel 60 Å F254 pre-coated plates (0.25 mm thickness). Visualization was accomplished by irradiation with a UV lamp and staining with I2 on silica gel. The elemental analysis was carried out with vario MICRO V1.3.2 Elemental Analyser system, GmbH instruments.General method A: typical experimental procedure for the synthesis of E/Z mixture of 2-benzoyl-4-oxo-4-phenyl-but-2-enoic acid ethyl esters
To a solution of aryl β-ketoester (1, 1.0 equiv.) in DMSO (3 mL), I2 (0.5 equiv.) and Cu(OAc)2·H2O (0.2 equiv.) was added at room temperature and heated at 80 °C under the atmosphere of air. Progress of the reaction was monitored by TLC. Upon on completion, the reaction mixture was allowed to cool to room temperature and quenched with sodium thiosulfate water and ethyl acetate. The organic phase was separated, dried over Na2SO4, filtered and concentrated. The crude product was purified by silica gel column chromatography using hexane/ethyl acetate as eluent.General method B: typical experimental procedure for the synthesis of quinoxalines
To a solution of 2-benzoyl-4-oxo-4-phenyl-but-2-enoic acid ethyl ester (2, 1.0 equiv.) in DMSO, benzene-1,2-diamine (3, 1.1 equiv.) and I2 (0.2 equiv.) was added at room temperature and stirred. Progress of the reaction was monitored by TLC. Upon on completion, the reaction mixture was allowed to cool to room temperature and quenched with sodium thiosulfate water and ethyl acetate. The organic phase was separated, dried over Na2SO4, filtered and concentrated. The crude product was purified by silica gel column chromatography using hexane/ethyl acetate as eluent.(E/Z)-2-Benzoyl-4-oxo-4-phenyl-but-2-enoic acid ethyl ester (2a)10a
The reaction was carried out according to general method A using 3-oxo-3-phenyl-propionic acid ethyl ester (1a, 100 mg, 0.520 mmol), I2 (66.0 mg, 0.260 mmol), Cu(OAc)2·H2O (20.8 mg, 0.104 mmol) and DMSO (3 mL). Conditions: 80 °C, 10 h. The title compound 2a (125.1 mg, 78% yield) was obtained as yellow oil after passing through a silica gel column chromatography.(E:Z = 83:17); (E)-isomer: 1H NMR (400 MHz, CDCl3): δ 8.17 (s, 1H), 7.98 (d, J = 8.0 Hz, 2H), 7.93 (d, J = 8.0 Hz, 2H), 7.64–7.57 (m, 2H), 7.52–7.46 (m, 4H), 4.30 (q, J = 14.2 Hz, 2H), 1.24 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 193.4, 188.5, 163.7, 144.9, 136.3, 136.0, 134.2, 133.5, 130.2, 128.9, 128.9, 128.7, 128.6, 62.4, 13.9.
(E/Z)-2-(Biphenyl-4-carbonyl)-4-biphenyl-4-yl-4-oxo-but-2-enoic acid ethyl ester (2b)
The reaction was carried out according to general method A using 3-biphenyl-4-yl-3-oxo-propionic acid ethyl ester (1b, 100 mg, 0.372 mmol), I2 (47.3 mg, 0.186 mmol), Cu(OAc)2·H2O (14.9 mg, 0.074 mmol) and DMSO (3 mL). Conditions: 80 °C, 12 h. The title compound 2b (121.8 mg, 71% yield) was obtained as light yellow semi solid after passing through a silica gel column chromatography.(E:Z = 86:14); (E)-isomer: 1H NMR (400 MHz, CDCl3): δ 8.21 (s, 1H), 8.05 (d, J = 8.8 Hz, 2H), 7.99 (d, J = 8.4 Hz, 2H), 7.71–7.60 (m, 8H), 7.48–7.45 (m, 6H), 4.31 (q, J = 14.2 Hz, 2H), 1.25 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 193.1, 187.9, 163.8, 147.0, 146.3, 144.9, 140.0, 139.5, 135.0, 134.7, 133.5, 130.8, 130.1, 129.6, 129.2, 129.1, 129.0, 129.0, 128.6, 128.3, 127.6, 127.5, 127.4, 127.2, 62.6, 14.0. HRMS (ESI): m/z [M + Na]+ calcd for C31H24NaO4: 483.1567; found: 483.1728.
(E/Z)-2-(4-Methyl-benzoyl)-4-oxo-4-p-tolyl-but-2-enoic acid ethyl ester (2c)
The reaction was carried out according to general method A using 3-oxo-3-p-tolyl-propionic acid ethyl ester (1c, 100 mg, 0.484 mmol), I2 (61.5 mg, 0.242 mmol), Cu(OAc)2·H2O (19.4 mg, 0.096 mmol) and DMSO (3 mL). Conditions: 80 °C, 6 h. The title compound 2c (130.4 mg, 80% yield) was obtained as semi solid after passing through a silica gel column chromatography.(E:Z = 84:16); (E)-isomer: 1H NMR (400 MHz, CDCl3): δ 8.04 (s, 1H), 7.79 (d, J = 8.4 Hz, 2H), 7.72 (d, J = 8.0 Hz, 2H), 7.20–7.16 (m, 4H), 4.19 (q, J = 14.2 Hz, 2H), 2.34 (s, 3H), 2.32 (s, 3H), 1.14 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 193.2, 187.9, 163.9, 145.4, 144.6, 144.5, 133.9, 133.6, 133.4, 129.6, 129.5, 129.1, 128.7, 62.4, 21.8, 21.8, 13.9. HRMS (ESI): m/z [M + Na]+ calcd for C19H16NaO4: 359.1254; found: 359.1254.
(E/Z)-4-Oxo-2-(4-phenoxy-benzoyl)-4-(4-phenoxy-phenyl)-but-2-enoic acid ethyl ester (2d)
The reaction was carried out according to general method A using 3-(4-benzyloxy-phenyl)-3-oxo-propionic acid ethyl ester (1d, 100 mg, 0.335 mmol), I2 (44.6 mg, 0.175 mmol), Cu(OAc)2·H2O (14.0 mg, 0.070 mmol) and DMSO (3 mL). Conditions: 80 °C, 3 h. The title compound 2d (125.6 mg, 72% yield) was obtained as yellow oil after passing through a silica gel column chromatography.(E:Z = 77:23); (E)-isomer: 1H NMR (400 MHz, CDCl3): δ 7.99 (s, 1H), 7.85 (d, J = 8.8 Hz, 2H), 7.78 (d, J = 8.8 Hz, 2H), 7.34–7.30 (m, 10H), 6.93–6.90 (m, 4H), 5.05 (s, 2H), 5.02 (s, 2H), 4.18 (q, J = 14.4 Hz, 4H), 1.13 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 192.0, 186.9, 164.0, 163.6, 163.1, 144.0, 136.2, 135.9, 133.6, 132.3, 131.4, 130.9, 129.8, 129.6, 128.7, 128.7, 128.3, 128.2, 127.5, 127.5, 115.0, 114.8, 114.6, 70.3, 70.2, 62.3, 13.9. HRMS (ESI): m/z [M + Na]+ calcd for C33H28NaO6: 543.1778; found: 543.1972.
(E/Z)-2-(4-Methoxy-benzoyl)-4-(4-methoxy-phenyl)-4-oxo-but-2-enoic acid ethyl ester (2e)
The reaction was carried out according to general method A using 3-(4-methoxy-phenyl)-3-oxo-propionic acid ethyl ester (1e, 100 mg, 0.449 mmol), I2 (57.1 mg, 0.224 mmol), Cu(OAc)2·H2O (18.0 mg, 0.089 mmol) and DMSO (3 mL). Conditions: 80 °C, 5 h. The title compound 2e (134.2 mg, 81% yield) was obtained as semi solid after passing through a silica gel column chromatography.(E:Z = 91:9); (E)-isomer: 1H NMR (400 MHz, CDCl3): δ 8.17 (s, 1H), 8.02 (d, J = 8.4 Hz, 2H), 7.95 (d, J = 8.8 Hz, 2H), 7.02 (d, J = 3.2 Hz, 2H), 7.00 (d, J = 3.6 Hz, 2H), 4.35 (q, J = 14.2 Hz, 2H), 3.94 (s, 3H), 3.92 (s, 3H), 1.30 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 192.1, 186.8, 164.4, 164.0, 163.9, 144.0, 133.5, 131.4, 130.9, 129.5, 129.3, 114.2, 114.0, 62.3, 55.6, 55.5, 14.0. HRMS (ESI): m/z [M + Na]+ calcd for C21H20NaO6: 391.1152; found: 391.1152.
(E/Z)-2-(4-Fluoro-benzoyl)-4-(4-fluoro-phenyl)-4-oxo-but-2-enoic acid ethyl ester (2f)
The reaction was carried out according to general method A using 3-(4-fluoro-phenyl)-3-oxo-propionic acid ethyl ester (1f, 100 mg, 0.475 mmol), I2 (60.4 mg, 0.237 mmol), Cu(OAc)2·H2O (19.0 mg, 0.095 mmol) and DMSO (3 mL). Conditions: 80 °C, 5 h. The title compound 2f (118.0 mg, 72% yield) was obtained as yellow oil after passing through a silica gel column chromatography.(E:Z = 84:16); (E)-isomer: 1H NMR (400 MHz, CDCl3): δ 8.09 (s, 1H), 8.03–7.97 (m, 2H), 7.93–7.85 (m, 2H), 7.22–7.08 (m, 4H), 4.28 (q, J = 14.2 Hz, 2H), 1.22 (t, J = 7.2 Hz, t); 13C NMR (100 MHz, CDCl3): δ 191.8, 186.9, 167.8, 167.3, 165.2, 164.8, 163.5, 144.8, 133.2, 131.8, 131.7, 131.2, 131.1, 116.4, 116.2, 116.1, 115.9, 62.6, 13.9. HRMS (ESI): m/z [M + Na]+ calcd for C19H14F2NaO4: 367.0752; found: 367.0856.
(E/Z)-2-(4-Chloro-benzoyl)-4-(4-chloro-phenyl)-4-oxo-but-2-enoic acid ethyl ester (2g)
The reaction was carried out according to general method A using 3-(4-chloro-phenyl)-3-oxo-propionic acid ethyl ester (1g, 100 mg, 0.441 mmol), I2 (56.0 mg, 0.220 mmol), Cu(OAc)2·H2O (17.6 mg, 0.088 mmol) and DMSO (3 mL). Conditions: 80 °C, 6 h. The title compound 2g (123.1 mg, 74% yield) was obtained as light semi solid after passing through a silica gel column chromatography.(E:Z = 83:17); (E)-isomer: 1H NMR (400 MHz, CDCl3): δ 8.09 (s, 1H), 7.90 (d, J = 8.8 Hz, 2H), 7.83 (d, J = 8.8 Hz, 2H), 7.48–7.43 (m, 4H), 4.31–4.25 (m, 2H), 1.22 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 192.2, 187.2, 163.4, 145.0, 141.1, 140.2, 134.4, 134.2, 133.1, 130.8, 130.3, 129.9, 129.4, 129.2, 62.7, 13.9. HRMS (ESI): m/z [M + Na]+ calcd for C19H14Cl2NaO4: 399.0161; found: 399.0161.
(E/Z)-2-(4-Bromo-benzoyl)-4-(4-cbromo-phenyl)-4-oxo-but-2-enoic acid ethyl ester (2h)
The reaction was carried out according to general method A using 3-(4-bromo-phenyl)-3-oxo-propionic acid ethyl ester (1h, 100 mg, 0.368 mmol), I2 (46.8 mg, 0.184 mmol), Cu(OAc)2·H2O (14.7 mg, 0.073 mmol) and DMSO (3 mL). Conditions: 80 °C, 6 h. The title compound 2h (128.9 mg, 75% yield) was obtained as yellow oil after passing through a silica gel column chromatography.(E:Z = 80:20); (E)-isomer: 1H NMR (400 MHz, CDCl3): δ 8.07 (s, 1H), 7.81 (d, J = 8.8 Hz, 2H), 7.74 (d, J = 8.4 Hz, 2H), 7.64–7.60 (m, 4H), 4.30–4.24 (m, 2H), 1.22 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 192.3, 187.4, 163.3, 145.0, 134.8, 134.7, 133.1, 132.4, 132.1, 130.3, 129.5, 62.7, 13.9. HRMS (ESI): m/z [M + Na]+ calcd for C19H14Br2NaO4: 486.9151; found: 486.9151.
(E/Z)-2-(4-Iodo-benzoyl)-4-(4-iodo-phenyl)-4-oxo-but-2-enoic acid ethyl ester (2i)
The reaction was carried out according to general method A using 3-(4-iodo-phenyl)-3-oxo-propionic acid ethyl ester (1i, 100 mg, 0.314 mmol), I2 (39.9 mg, 0.157 mmol), Cu(OAc)2·H2O (12.5 mg, 0.062 mmol) and DMSO (3 mL). Conditions: 80 °C, 6 h. The title compound 2i (130.3 mg, 74% yield) was obtained as yellow oil after passing through a silica gel column chromatography.(E:Z = 95:5); (E)-isomer: 1H NMR (400 MHz, CDCl3): δ 8.05 (s, 1H), 7.86–7.81 (m, 4H), 7.64 (d, J = 8.4 Hz, 2H), 7.58 (d, J = 8.4 Hz, 2H), 4.27 (q, J = 14.0 Hz, 2H), 1.21 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 192.6, 187.7, 163.3, 145.0, 138.4, 138.1, 135.3, 135.1, 133.0, 130.1, 129.8, 103.0, 101.9, 62.7, 13.9. HRMS (ESI): m/z [M + Na]+ calcd for C19H14I2NaO4: 582.8874; found: 582.8874.
(E/Z)-2-(4-Nitro-benzoyl)-4-(4-nitro-phenyl)-4-oxo-but-2-enoic acid ethyl ester (2j)
The reaction was carried out according to general method A using 3-(4-nitro-phenyl)-3-oxo-propionic acid ethyl ester (1j, 100 mg, 0.421 mmol), I2 (53.5 mg, 0.21 mmol), Cu(OAc)2·H2O (16.8 mg, 0.084 mmol) and DMSO (3 mL). Conditions: 80 °C, 8 h. The title compound 2j (115.9 mg, 69% yield) was obtained as semi solid after passing through a short silica gel column chromatography.(E:Z = 78:22); (E)-isomer: 1H NMR (400 MHz, CDCl3): δ 8.30–8.26 (m, 4H), 8.09 (s, 1H), 8.06 (d, J = 9.2 Hz, 2H), 7.99 (d, J = 9.2 Hz, 2H), 4.25 (q, J = 14.4 Hz, 2H), 1.17 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 191.5, 187.1, 162.7, 151.0, 150.7, 145.8, 140.0, 140.0, 132.8, 130.0, 129.4, 124.3, 124.1, 63.2, 13.9. HRMS (ESI): m/z [M + Na]+ calcd for C19H14N2NaO8: 421.0648; found: 421.0648.
(E/Z)-2-(3-Chloro-benzoyl)-4-(3-chloro-phenyl)-4-oxo-but-2-enoic acid ethyl ester (2k)
The reaction was carried out according to general method A using 3-(3-chloro-phenyl)-3-oxo-propionic acid ethyl ester (1k, 100 mg, 0.441 mmol), I2 (56.0 mg, 0.220 mmol), Cu(OAc)2·H2O (17.6 mg, 0.088 mmol) and DMSO (3 mL). Conditions: 80 °C, 8 h. The title compound 2k (109.8 mg, 66% yield) was obtained as yellow oil after passing through a silica gel column chromatography.(E:Z = 80:20); (E/Z)-mixture: 1H NMR (400 MHz, CDCl3): δ 8.08 (s, 1H), 7.99–7.97 (m, 1H), 7.92–7.91 (m, 1H), 7.87–7.83 (m, 1H), 7.75–7.71 (m, 1H), 7.60–7.53 (m, 1H), 7.44–7.38 (m, 2H), 4.32–4.25 (m, 2H), 1.24–1.21 (m, 3H); 13C NMR (100 MHz, CDCl3): δ 191.9, 189.6, 189.2, 187.2, 163.2, 162.8, 162.1, 145.1, 142.2, 142.0, 137.5, 137.3, 137.2, 136.8, 135.4, 135.3, 135.2, 135.1, 134.7, 134.3, 134.2, 133.9, 133.8, 133.6, 133.1, 131.0, 130.3, 130.2, 130.1, 129.8, 128.9, 128.9, 128.4, 128.3, 127.5, 127.1, 127.0, 126.6, 62.9, 62.8, 13.9, 13.7. HRMS (ESI): m/z [M + Na]+ calcd for C19H14Cl2NaO4: 399.0161; found: 399.0161.
(E/Z)-2-(3-Methoxy-benzoyl)-4-(3-methoxy-phenyl)-4-oxo-but-2-enoic acid ethyl ester (2l)
The reaction was carried out according to general method A using 3-(3-methoxy-phenyl)-3-oxo-propionic acid ethyl ester (1l, 100 mg, 0.449 mmol), I2 (57.1 mg, 0.224 mmol), Cu(OAc)2·H2O (18.0 mg, 0.089 mmol) and DMSO (3 mL). Conditions: 80 °C, 8 h. The title compound 2l (112.7 mg, 68% yield) was obtained as yellow semi solid after passing through a silica gel column chromatography.(E:Z = 80:20); (E)-isomer: 1H NMR (400 MHz, CDCl3): δ 8.03 (s, 1H), 7.52–7.50 (m, 1H), 7.45–7.44 (m, 1H), 7.36–7.30 (m, 3H), 7.26 (t, J = 7.8 Hz, 1H), 7.09–7.03 (m, 2H), 4.20 (q, J = 14.2 Hz, 2H), 3.78 (s, 3H), 3.73 (s, 3H), 1.15 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 193.2, 188.1, 163.7, 160.1, 159.9, 144.9, 137.6, 137.1, 133.3, 129.9, 129.8, 121.7, 121.6, 121.3, 120.5, 112.5, 112.0, 62.5, 55.5, 55.4, 13.9. HRMS (ESI): m/z [M + Na]+ calcd for C21H20NaO6: 391.1152; found: 391.1311.
(E/Z)-2-(2-Methoxy-benzoyl)-4-(2-methoxy-phenyl)-4-oxo-but-2-enoic acid ethyl ester (2m)
The reaction was carried out according to general method A using 3-(3-methoxy-phenyl)-3-oxo-propionic acid ethyl ester (1m, 100 mg, 0.449 mmol), I2 (57.1 mg, 0.224 mmol), Cu(OAc)2·H2O (18.0 mg, 0.089 mmol) and DMSO (3 mL). Conditions: 80 °C, 12 h. The title compound 2m (112.7 mg, 68% yield) was obtained as yellow oil after passing through a silica gel column chromatography.(E:Z = 88:12); (E)-isomer: 1H NMR (400 MHz, CDCl3): δ 8.06 (d, J = 8.0 Hz, 1H), 7.78 (s, 1H), 7.59 (d, J = 7.6 Hz, 1H), 7.58–7.42 (m, 2H), 7.05 (t, J = 7.8 Hz, 1H), 6.92 (t, J = 6.8 Hz, 3H), 4.25 (q, J = 14.2 Hz, 2H), 3.89 (s, 3H), 3.76 (s, 3H), 1.19 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 191.6, 190.4, 164.4, 159.5, 159.3, 145.2, 134.8, 134.7, 134.4, 131.0, 130.1, 127.1, 126.1, 121.1, 120.8, 112.1, 111.8, 61.5, 55.8, 55.5, 14.0. HRMS (ESI): m/z [M + Na]+ calcd for C21H20NaO6: 391.1152; found: 391.1231.
(E/Z)-2-(3,4-Dimethyl-benzoyl)-4-(3,4-dimethyl-phenyl)-4-oxo-but-2-enoic acid ethyl ester (2n)
The reaction was carried out according to general method A using 3-(4-nitro-phenyl)-3-oxo-propionic acid ethyl ester (1n, 100 mg, 0.454 mmol), I2 (57.6 mg, 0.227 mmol), Cu(OAc)2·H2O (18.1 mg, 0.09 mmol) and DMSO (3 mL). Conditions: 80 °C, 5.5 h. The title compound 2n (125.7 mg, 76% yield) was obtained as semi solid after passing through a short silica gel column chromatography.(E:Z = 92:8); (E)-isomer: 1H NMR (400 MHz, CDCl3): δ 8.04 (s, 1H), 7.65–7.62 (m, 3H), 7.51 (dd, J = 1.6 Hz, 1.6 Hz, 1H), 7.14 (d, J = 8.4 Hz, 1H), 7.10 (d, J = 8.0 Hz, 1H), 4.20 (q, J = 14.4 Hz, 4H), 2.23 (s, 3H), 2.21 (s, 9H), 1.15 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 193.4, 188.0, 164.1, 144.6, 144.1, 143.3, 137.4, 137.1, 134.2, 133.9, 133.5, 130.1, 130.0, 130.0, 129.4, 126.7, 126.5, 62.4, 20.2, 20.2, 19.8, 19.7, 14.0. HRMS (ESI): m/z [M + Na]+ calcd for C23H24NaO4: 387.1572; found: 387.1572.
(E/Z)-4-Oxo-2-(thiophene-2-carbonyl)-4-thiophen-2-yl-but-2-enoic acid ethyl ester (2o)
The reaction was carried out according to general method A using 3-oxo-3-thiophen-2-yl-propionic acid ethyl ester (1o, 100 mg, 0.504 mmol), I2 (64.0 mg, 0.252 mmol), Cu(OAc)2·H2O (20.1 mg, 0.1 mmol) and DMSO (3 mL). Conditions: 80 °C, 3 h. The title compound 2o (126.0 mg, 78% yield) was obtained as yellow oil after passing through a short silica gel column chromatography.(E:Z = 78:22); (E)-isomer: 1H NMR (400 MHz, CDCl3): δ 7.93 (s, 1H), 7.88 (dd, J = 0.8 Hz, 1.0 Hz, 1H), 7.75 (dd, J = 1.2 Hz, 0.8 Hz, 1H), 7.69 (dd, J = 1.2 Hz, 0.8 Hz, 1H), 7.19 (t, J = 4.4 Hz, 3H), 7.09 (t, J = 4.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 184.9, 180.0, 163.4, 144.1, 143.4, 143.0, 136.4, 134.5, 133.9, 133.3, 133.1, 128.7, 128.2, 62.6, 13.9. HRMS (ESI): m/z [M + Na]+ calcd for C15H12NaO4S2: 343.0069; found: 343.0152.
(E/Z)-2-(Naphthalene-1-carbonyl)-4-naphthalen-1-yl-4-oxo-but-2-enoic acid ethyl ester (2p)
The reaction was carried out according to general method A using 3-naphthalen-1-yl-3-oxo-propionic acid ethyl ester (1p, 100 mg, 0.412 mmol), I2 (52.4 mg, 0.206 mmol), Cu(OAc)2·H2O (16.5 mg, 0.082 mmol) and DMSO (3 mL). Conditions: 80 °C, 6 h. The title compound 2p (126.4 mg, 75% yield) was obtained as yellow oil after passing through a silica gel column chromatography.(E:Z = 90:10); (E)-isomer: 1H NMR (400 MHz, CDCl3): δ 8.99 (d, J = 8.8 Hz, 2H), 8.35 (d, J = 8.4 Hz, 2H), 8.05 (s, 1H), 8.02–7.99 (m, 3H), 7.95 (d, J = 7.2 Hz, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.81 (d, J = 8.0 Hz, 1H), 7.62 (t, J = 7.8 Hz, 3H), 7.55 (t, J = 6.6, 3H), 7.42–7.51 (m, 3H), 4.31 (q, J = 14.2 Hz, 2H), 1.21 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 194.3, 192.3, 164.3, 144.3, 137.3, 134.3, 133.9, 131.5, 130.4, 128.5, 128.4, 128.4, 128.2, 126.7, 126.6, 126.1, 125.5, 124.2, 124.1, 62.3, 13.9. HRMS (ESI): m/z [M + Na]+ calcd for C27H20NaO4: 431.1254; found: 431.1386.
(E/Z)-2-(4-Methoxy-benzoyl)-4-(4-methoxy-phenyl)-4-oxo-but-2-enoic acid methyl ester (2q)
The reaction was carried out according to general method A using 3-oxo-3-phenyl-propionic acid methyl ester (1q, 100 mg, 0.480 mmol), I2 (60.9 mg, 0.240 mmol), Cu(OAc)2·H2O (19.1 mg, 0.096 mmol) and DMSO (3 mL). Conditions: 80 °C, 5.5 h. The title compound 2q (136.1 mg, 80% yield) was obtained as semi solid after passing through a short silica gel column chromatography.(E:Z = 92:8); (E)-isomer: 1H NMR (400 MHz, CDCl3): δ 8.12 (s, 1H), 7.96 (d, J = 8.8 Hz, 2H), 7.88 (d, J = 8.8 Hz, 2H), 6.96–6.93 (m, 4H), 3.88 (s, 3H), 3.85 (s, 3H), 3.81 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 192.0, 186.6, 164.6, 164.5, 164.0, 143.5, 133.7, 131.4, 131.0, 129.5, 129.2, 114.2, 114.1, 55.6, 55.5, 53.2. HRMS (ESI): m/z [M + Na]+ calcd for C20H18O6: 377.0995; found: 377.1115.
2-Phenyl-quinoxaline (4a)24
The reaction was carried out according to general method B using 2-benzoyl-4-oxo-4-phenyl-but-2-enoic acid ethyl ester (2a, 100 mg, 0.324 mmol), benzene-1,2-diamine (3a 38.6 mg, 0.356 mmol), I2 (16.5 mg, 0.064 mmol) and DMSO (2 mL). Conditions: r.t., 3 h. The title compound 4a (57.5 mg, 86% yield) was obtained as white crystal after passing through a short silica gel column chromatography.Mp: 71–73 °C; 1H NMR (400 MHz, CDCl3): δ 9.33 (s, 1H), 8.21–8.11 (m, 4H), 7.81–7.72 (m, 2H), 7.59–7.50 (m, 3H); 13C NMR (100 MHz, CDCl3): δ 151.9, 143.3, 142.4, 141.6, 136.8, 130.3, 130.2, 129.7, 129.6, 129.2, 129.1, 127.6.
2-p-Tolyl-quinoxaline (4b)24
The reaction was carried out according to general method B using 2-(4-methyl-benzoyl)-4-oxo-4-m-tolyl-but-2-enoic acid ethyl ester (2c, 100 mg, 0.297 mmol), benzene-1,2-diamine (3a, 35.4, 0.327 mmol), I2 (15.1 mg, 0.059 mmol) and DMSO (2 mL). Conditions: r.t., 3 h. The title compound 4b (55.0 mg, 84% yield) was obtained as yellow solid after passing through a short silica gel column chromatography.Mp: 90–92 °C; 1H NMR (400 MHz, CDCl3): δ 9.24 (s, 1H), 8.09–8.03 (m, 4H), 7.73–7.64 (m, 2H), 7.30 (d, J = 8.0 Hz, 2H), 2.38 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 151.9, 143.1, 142.3, 141.2, 140.6, 133.9, 130.3, 129.9, 129.5, 129.4, 129.0, 127.5, 21.4.
2-(3-Methoxy-phenyl)-quinoxaline (4c)25
The reaction was carried out according to general method B using 2-(4-methoxy-benzoyl)-4-(3-methoxy-phenyl)-4-oxo-but-2-enoic acid ethyl ester (2e, 100 mg, 0.271 mmol), benzene-1,2-diamine (3a, 32.6 mg, 0.298 mmol), I2 (13.8 mg, 0.054 mmol) and DMSO (2 mL). Conditions: r.t., 3 h. The title compound 4c (52.6 mg, 82% yield) was obtained as light yellow solid after passing through a short silica gel column chromatography.Mp: 108–110 °C; 1H NMR (400 MHz, CDCl3): δ 9.21 (s, 1H), 8.07–8.01 (m, 2H), 7.70–7.62 (m, 4H), 7.37 (t, J = 8.0 Hz, 1H), 6.96 (dd, J = 8.4, 2.4 Hz, 1H), 3.84 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 160.3, 151.6, 143.4, 142.2, 141.7, 138.2, 130.3, 130.2, 129.6, 129.6, 129.1, 119.9, 116.2, 112.7, 55.5.
2-(4-Fluoro-phenyl)-quinoxaline (4d)25
The reaction was carried out according to general method B using 2-(4-fluoro-benzoyl)-4-(3-fluoro-phenyl)-4-oxo-but-2-enoic acid ethyl ester (2f, 100 mg, 0.29 mmol), benzene-1,2-diamine (3a, 34.5 mg, 0.319 mmol), I2 (14.7 mg, 0.058 mmol) and DMSO (2 mL). Conditions: r.t., 2.5 h. The title compound 4d (55.3 mg, 85% yield) was obtained as white crystal after passing through a short silica gel column chromatography.Mp: 97–99 °C; 1H NMR (400 MHz, CDCl3): δ 9.30 (s, 1H), 8.21 (d, J = 6.4 Hz, 2H), 8.13 (d, J = 7.8 Hz, 2H), 7.81–7.74 (m, 2H), 7.26 (d, J = 8.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 165.5, 163.1, 150.8, 142.9, 142.2, 141.5, 133.0, 130.4, 129.6, 129.6, 129.6, 129.5, 129.1, 116.4, 116.1.
2-(4-Chloro-phenyl)-quinoxaline (4e)24
The reaction was carried out according to general method B using 2-(4-chloro-benzoyl)-4-(3-chloro-phenyl)-4-oxo-but-2-enoic acid ethyl ester (2g, 100 mg, 0.265 mmol), benzene-1,2-diamine (3a, 31.5 mg, 0.291 mmol), I2 (13.5 mg, 0.053 mmol) and DMSO (2 mL). Conditions: r.t., 3.0 h. The title compound 4e (51.0 mg, 80% yield) was obtained as light yellow crystal after passing through a short silica gel column chromatography.Mp: 130–132 °C; 1H NMR (400 MHz, CDCl3): δ 9.29 (s, 1H), 8.16–8.11 (m, 4H), 7.81–7.73 (m, 2H), 7.53 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 150.6, 142.8, 142.2, 141.7, 136.6, 135.2, 130.5, 129.8, 129.6, 129.2, 128.8.
2-Biphenyl-4-yl-quinoxaline (4f)24
The reaction was carried out according to general method B using 2-(biphenyl-4-carbonyl)-4-biphenyl-4-yl-4-oxo-but-2-enoic acid ethyl ester (2b, 100 mg, 0.217 mmol), benzene-1,2-diamine (3a, 25.8 mg, 0.238 mmol), I2 (11.0 mg, 0.043 mmol) and DMSO (2 mL). Conditions: r.t., 4.5 h. The title compound 4f (50.3 mg, 82% yield) was obtained as grey solid after passing through a short silica gel column chromatography.Mp: 131–133 °C; 1H NMR (400 MHz, CDCl3): δ 9.39 (s, 1H), 8.30 (d, J = 7.6 Hz, 2H), 8.16 (dd, J = 8.4 Hz, 8.0 Hz, 2H), 7.82–7.74 (m, 4H), 7.69 (d, J = 7.6 Hz, 2H), 7.49 (t, J = 7.4 Hz, 2H), 7.40 (t, J = 7.2 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 151.5, 143.2, 143.1, 142.4, 141.6, 140.3, 135.6, 130.3, 129.6, 129.5, 129.1, 128.9, 128.0, 127.9, 127.2.
2-Naphthalen-1-yl-quinoxaline (4g)24
The reaction was carried out according to general method B using 2-(naphthalene-1-carbonyl)-4-naphthalen-1-yl-4-oxo-but-2-enoic acid ethyl ester (2p, 100 mg, 0.244 mmol), benzene-1,2-diamine (3a, 29.1 mg, 0.269 mmol), I2 (12.4 mg, 0.048 mmol) and DMSO (2 mL). Conditions: r.t., 4.0 h. The title compound 4g (48.3 mg, 77% yield) was obtained as grey solid after passing through a short silica gel column chromatography.Mp: 135–136 °C; 1H NMR (400 MHz, CDCl3): δ 9.09 (s, 1H), 8.16–8.08 (m, 3H), 7.95–7.88 (m, 2H), 7.78–7.69 (m, 3H), 7.56 (t, J = 7.6 Hz, 1H), 7.50–7.43 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 154.3, 146.6, 142.2, 141.4, 135.1, 134.1, 131.2, 130.4, 130.2, 130.0, 129.7, 129.3, 128.7, 128.5, 127.2, 126.4, 125.4, 125.1.
2-Thiophen-2-yl-quinoxaline (4h)24
The reaction was carried out according to general method B using 4-oxo-2-(thiophene-2-carbonyl)-4-thiophen-2-yl-but-2-enoic acid ethyl ester (2o, 100 mg, 0.312 mmol), benzene-1,2-diamine (3a, 37.1 mg, 0.343 mmol), I2 (15.8 mg, 0.062 mmol) and DMSO (2 mL). Conditions: r.t., 2.5 h. The title compound 4h (54.3 mg, 82% yield) was obtained as yellow crystal after passing through a short silica gel column chromatography.Mp: 114–116 °C; 1H NMR (400 MHz, CDCl3): δ 9.24 (s, 1H), 8.08–8.05 (dd, J = 3.6 Hz, 3.2 Hz, 2H), 7.86 (d, J = 3.6 Hz, 1H), 7.76–7.67 (m, 2H), 7.55 (d, J = 4.8 Hz, 1H), 7.20 (t, J = 4.4 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 147.4, 142.3, 142.2, 142.1, 141.4, 130.4, 129.8, 129.2, 129.1, 128.5, 127.0.
6-Methyl-2-pheny-quinoxaline and 7-methyl-2-pheny-quinoxaline (4i)24
The reaction was carried out according to general method B using 2-benzoyl-4-oxo-4-phenyl-but-2-enoic acid ethyl ester (2a, 100 mg, 0.324 mmol), 4-methyl-benzene-1,2-diamine (3b, 43.6 mg, 0.356 mmol), I2 (16.5 mg, 0.064 mmol) and DMSO (2 mL). Conditions: r.t., 3.0 h. The title compound 4i (56.4 mg, 79% yield) was obtained as white crystal after passing through a short silica gel column chromatography.Mp: 108–110 °C; 1H NMR (400 MHz, CDCl3): δ 9.28–9.25 (m, 1H), 8.18 (d, J = 7.2 Hz, 2H), 8.05–7.99 (m, 1H), 7.93–7.89 (m, 1H), 7.62–7.50 (m, 4H), 2.61 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 151.8, 151.1, 143.1, 142.4, 141.6, 140.8, 140.8, 140.2, 140.1, 137.0, 132.6, 131.9, 130.1, 130.0, 129.2, 129.1, 128.6, 128.5, 127.9, 127.5, 127.4, 21.8, 21.8.
2-(4-Methoxy-phenyl)-6-methyl-quinoxaline and 2-(4-methoxy-phenyl)-6-methyl-quinoxaline (4j)
The reaction was carried out according to general method B using 2-(4-methoxy-benzoyl)-4-(3-methoxy-phenyl)-4-oxo-but-2-enoic acid ethyl ester (2e, 100 mg, 0.271 mmol), 4-methyl-benzene-1,2-diamine (3b, 13.7 mg, 0.298 mmol), I2 (11.2 mg, 0.054 mmol) and DMSO (2 mL). Conditions: r.t., 3.0 h. The title compound 4j (55.0 mg, 81% yield) was obtained as yellow solid after passing through a short silica gel column chromatography.Mp: 121–123 °C; 1H NMR (400 MHz, CDCl3): δ 9.24–9.21 (m, 1H), 8.18–8.13 (m, 2H), 8.02–7.96 (m, 1H), 7.89–7.86 (m, 1H), 7.60–7.53 (m, 1H), 7.07 (d, J = 8.8 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 161.5, 161.4, 151.4, 150.8, 142.8, 142.1, 141.1, 140.8, 139.6, 132.5, 131.4, 129.5, 128.9, 128.9, 128.8, 128.5, 128.2, 127.9, 114.6, 55.4, 21.8, 21.8. HRMS (ESI): m/z [M + H]+ calcd for C16H15N2O: 251.1184; found: 251.1201.
2-(3-Chloro-phenyl)-pyrazine (6)
To a solution of 2-(3-chloro-benzoyl)-4-(3-chloro-phenyl)-4-oxo-but-2-enoic acid ethyl ester (2j, 100 mg, 0.265 mmol) in DMSO (3 mL), ethane-1,2-diamine (5, 17.5 mg, 0.291 mmol), I2 (13.5 mg, 0.05 mmol) and K2CO3 (44 mg, 0.318 mmol) was added at room temperature and heated at 80 °C for 8 h. After completion, the reaction mixture was allowed to cool to room temperature and quenched with sodium thiosulfate water and ethyl acetate. The organic phase was separated, dried over Na2SO4, filtered and concentrated. The crude product was purified by silica gel column chromatography using hexane/ethyl acetate as eluent. The title compound 6 (36.4 mg, 72% yield) was obtained as light yellow solid after passing through a short silica gel column chromatography.Mp: 92–94 °C; 1H NMR (400 MHz, CDCl3): δ 8.95 (s, 1H), 8.58 (dd, J = 2.0, 1.6 Hz, 1H), 8.48 (d, J = 2.4 Hz, 1H), 7.98 (s, 1H), 7.83–7.81 (m, 1H), 7.39–7.38 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 151.4, 144.3, 143.6, 142.2, 138.1, 135.3, 130.3, 130.0, 127.2, 124.9. HRMS (ESI): m/z [M + H]+ calcd for C10H7ClN2Na: 213.0195; found: 213.0198.
(9,9a-Dihydro-4aH-β-carbolin-1-yl)-(4-methoxy-phenyl)-methanone (8)21
To a solution of 2-(4-methoxy-benzoyl)-4-(4-methoxy-phenyl)-4-oxo-but-2-enoic acid ethyl ester (2j, 100 mg, 0.271 mmol) in DMSO (3 mL), tryptamine (7, 47.8 mg, 0.298 mmol), and Cu(OAc)2·H2O (16.3 mg, 0.081 mmol) was added at room temperature and heated at 80 °C for 6 h. After completion, the reaction mixture was allowed to cool to room temperature and quenched with water and ethyl acetate. The organic phase was separated, dried over Na2SO4, filtered and concentrated. The crude product was purified by silica gel column chromatography using hexane/ethyl acetate as eluent. The title compound 8 (62.4 mg 76% yield) was obtained as light brown solid after passing through a short silica gel column chromatography.Mp: 174–176 °C; 1H NMR (400 MHz, CDCl3): δ 10.48 (s, 1H), 8.63 (d, J = 5.2 Hz, 1H), 8.48 (d, J = 7.0 Hz, 2H), 8.21–8.18 (m, 2H), 7.64–7.63 (m, 2H), 7.39–7.35 (m, 1H), 7.07 (dd, J = 2.0, 2.0 Hz, 2H), 3.94 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 193.5, 163.3, 141.0, 137.8, 137.3, 137.0, 133.8, 131.6, 130.3, 129.2, 121.8, 120.9, 120.6, 118.2, 113.4, 112.0, 55.5.
Acknowledgements
G.S. thanks UGC, New Delhi, for the award of a fellowship. We thank DST-FIST for the use of instrument facility at the school of chemistry, Bharathidasan University.Notes and references
- (a) L. F. Tietze, Chem. Rev., 1996, 96, 115 CrossRef CAS PubMed; (b) H. Clavier and H. Pellissier, Adv. Synth. Catal., 2012, 354, 3347 CrossRef CAS PubMed.
- For self-sorting domino reactions: (a) G. Yin, B. Zhou, X. Meng, A. X. Wu and Y. Pan, Org. Lett., 2006, 8, 2245 CrossRef CAS PubMed; (b) G. Yin, Z. Wang, A. Chen, M. Gao, A. X. Wu and Y. Pan, J. Org. Chem., 2008, 73, 3377 CrossRef CAS PubMed; (c) M. Gao, G. Yin, Z. Wang, Y. Wu, C. Guo, Y. Pan and A. X. Wu, Tetrahedron, 2009, 65, 6047 CrossRef CAS PubMed.
- (a) J. S. Webb, D. B. Cosulich, J. H. Mowat, J. B. Patrick, R. W. Broschard, W. E. Meyer, R. P. Williams, C. F. Wolf, W. Fulmor, C. Pidacs and J. E. Lancaster, J. Am. Chem. Soc., 1962, 84, 3185 CrossRef CAS; (b) J. Salva and D. J. Faulkner, J. Org. Chem., 1990, 55, 1941 CrossRef CAS.
- For synthetic applications of 2-carboalkoxy 1,4-enediones: (a) M. Gao, Y. Yang, Y. D. Wu, C. Deng, W. M. Shu, D. X. Zhang, L. P. Cao, N. F. She and A. X. Wu, Org. Lett., 2010, 12, 4026 CrossRef CAS PubMed; (b) Y. Yang, M. Gao, L. M. Wu, C. Deng, D. X. Zhang, Y. Gao, Y. P. Zhu and A. X. Wu, Tetrahedron, 2011, 67, 5142 CrossRef CAS PubMed; (c) Q. Gao, Y. Zhu, M. Lian, M. Liu, J. Yuan, G. Yin and A. X. Wu, J. Org. Chem., 2012, 77, 9865 CrossRef CAS PubMed; (d) Y. Yang, W. M. Shu, S. B. Yu, M. Gao and A. X. Wu, J. Org. Chem., 2013, 78, 5418 CrossRef CAS PubMed; (e) Y. Yang, W. M. Shu, S. B. Yu, F. Ni, M. Gao and A. X. Wu, Chem. Commun., 2013, 49, 1729 RSC; (f) W. M. Shu, Y. Yang, D. X. Zhang, L. M. Wu, Y. P. Zhu, G. D. Yin and A. X. Wu, Org. Lett., 2013, 15, 456 CrossRef CAS PubMed; (g) Y. Yang, F. Ni, W. M. Shu and A. X. Wu, Tetrahedron, 2014, 70, 6733 CrossRef CAS PubMed.
- (a) C. Asta, J. Conrad, S. Mika and U. Beifuss, Green Chem., 2011, 13, 3066 RSC; (b) M. Nandakumar, R. Sivasakthikumaran and A. K. Mohanakrishnan, Eur. J. Org. Chem., 2012, 3647 CrossRef CAS PubMed.
- W. Baratta, A. D. Zotto and P. Rigo, Chem. Commun., 1997, 2163 RSC.
- J. Q. Yu and E. J. Corey, J. Am. Chem. Soc., 2003, 125, 3232 CrossRef CAS PubMed.
- B. Crone and S. F. Kirsch, Chem. Commun., 2006, 764 RSC.
- K. Xu, Y. Fang, Z. Yan, Z. Zha and Z. Wang, Org. Lett., 2013, 15, 2148 CrossRef CAS PubMed.
- For synthesis of unsymmetrical 1,4-enediones: (a) M. Gao, Y. Yang, Y. D. Wu, C. Deng, L. P. Cao, X. G. Meng and A. X. Wu, Org. Lett., 2010, 12, 1856 CrossRef CAS PubMed; (b) Y. P. Zhu, Q. Cai, Q. H. Gao, F. C. Jia, M. C. Liu, M. Gao and A. X. Wu, Tetrahedron, 2013, 69, 6392 CrossRef CAS PubMed.
- For reviews of C–C bond activation, see: (a) C. H. Jun, Chem. Soc. Rev., 2004, 33, 610 RSC, for reference, see: (b) A. M. Dreis and C. J. Douglas, J. Am. Chem. Soc., 2009, 131, 412 CrossRef CAS PubMed; (c) M. P. Watson and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 12594 CrossRef CAS PubMed; (d) Y. Nakao, S. Ebata, A. Yada, T. Hiyama, M. Ikawa and S. Ogoshi, J. Am. Chem. Soc., 2008, 130, 12874 CrossRef CAS PubMed; (e) A. Wilsily, Y. Nguyen and E. J. Fillion, J. Am. Chem. Soc., 2009, 131, 15606 CrossRef CAS PubMed.
- (a) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed; (b) A. de Meijere, Chem. Rev., 2003, 103, 931 CrossRef CAS; (c) M. Lin, F. Li, L. Jiao and Z.-X. Yu, J. Am. Chem. Soc., 2011, 133, 1690 CrossRef CAS PubMed; (d) T. Seiser and N. Cramer, J. Am. Chem. Soc., 2010, 132, 5340 CrossRef CAS PubMed.
- (a) P. Grubmuller, W. F. Maier, P. V. R. Schleyer, M. A. McKervey and J. J. Rooney, Chem. Ber., 1980, 113, 1989 CrossRef PubMed; (b) W. F. Maier, P. Grubmuller, I. Thies, P. M. Stein, M. A. McKervey and P. V. R. Schleyer, Angew. Chem., Int. Ed., 1979, 18, 939 CrossRef PubMed.
- (a) D. Necas, M. Tursky and M. Kotora, J. Am. Chem. Soc., 2004, 126, 10222 CrossRef CAS PubMed; (b) Q. Xu, B. Cheng, X. Ye and H. Zhai, Org. Lett., 2009, 11, 4136 CrossRef CAS PubMed; (c) H. Li, W. Li, W. Liu, Z. He and Z. Li, Angew. Chem., Int. Ed., 2011, 50, 2975 CrossRef CAS PubMed; (d) W. Li, X. Zheng and Z. Li, Adv. Synth. Catal., 2013, 355, 181 CrossRef CAS PubMed; (e) Y. Yang, F. Ni, W. M. Shu and A. X. Wu, Chem.–Eur. J., 2014, 20, 11776 CrossRef CAS PubMed.
- (a) A. Ilangovan and G. Satish, Org. Lett., 2013, 15, 5726 CrossRef CAS PubMed; (b) A. Ilangovan and G. Satish, J. Org. Chem., 2014, 79, 4984 CrossRef CAS PubMed.
- (a) A. P. Krapcho, ARKIVOC, 2007, 1 CrossRef CAS; (b) A. P. Krapcho, ARKIVOC, 2007, 54 CrossRef CAS.
- (a) G. Arthur, K. B. Elor, G. S. Robert, Z. Z. Guo, J. P. Richard, D. Stanley, R. K. John and T. Sean, J. Med. Chem., 2005, 48, 744 CrossRef PubMed; (b) E. S. Lainne, J. S. William and C. R. Robert, J. Med. Chem., 2002, 45, 5604 CrossRef PubMed; (c) J. Andres, Z. Belen, A. Ibnacio and M. Antonio, J. Med. Chem., 2005, 48, 2019 CrossRef PubMed; (d) B. Zarranz, A. Jaso, I. Aldana and A. Monge, Bioorg. Med. Chem., 2004, 12, 3711 CrossRef CAS PubMed; (e) K. M. Amin, M. M. F. Ismail, E. Noaman, D. H. Soliman and Y. A. Ammar, Bioorg. Med. Chem., 2006, 14, 6917 CrossRef CAS PubMed; (f) H. Xu and L. L. Fan, Eur. J. Med. Chem., 2011, 46, 1919 CrossRef CAS PubMed; (g) G. K. Rao, R. B. Kotnal and T. Pai, J. Chem. Pharm. Res., 2010, 2, 368 CAS; (h) J. Guillon, S. Moreau, E. Mouray, V. Sinou, I. Forfar, S. B. Fabre, V. Desplat, P. Millet, D. Parzy, C. Jarry and P. Grellier, Bioorg. Med. Chem., 2008, 16, 9133 CrossRef CAS PubMed.
- (a) S. Dailey, J. W. Feast, R. J. Peace, I. C. Sage, S. Till and E. L. Wood, Mater. Chem., 2001, 11, 2238 RSC; (b) D. O'Brien, M. S. Weaver, D. G. Lidzey and D. D. C. Bradley, Appl. Phys. Lett., 1996, 69, 881 CrossRef PubMed.
- K. R. Justin Thomas, M. Velusamy, T. L. Jiann, C. C. Hao and T. Yu, Chem. Mater., 2005, 17, 1860 CrossRef.
- E. D. Brock, D. M. Lewis, T. I. Yousaf and H. H. Harper, WO 9951688, The Procter & Gamble, USA, 1999.
- H. Jin, P. Zhang, K. Bijian, S. Ren, S. Wan, M. A. Alaoui-Jamali and T. Jiang, Mar. Drugs, 2013, 11, 1427 CrossRef CAS PubMed.
- Recent papers for radical formation: (a) M. T. Bovino, T. W. Liwosz, N. E. Kendel, Y. Miller, N. Tyminska, E. Zurek and S. R. Chemler, Angew. Chem., Int. Ed., 2014, 53, 6383 CrossRef CAS PubMed; (b) C. M. McMahon and E. J. Alexanian, Angew. Chem., Int. Ed., 2014, 53, 5974 CrossRef CAS PubMed; (c) J.-H. Fan, W.-T. Wei, M.-B. Zhou, R.-J. Song and J.-H. Li, Angew. Chem., Int. Ed., 2014, 53, 6650 CrossRef CAS PubMed; (d) T. Nishikata, Y. Noda, R. Fujimoto and T. Sakashita, J. Am. Chem. Soc., 2013, 135, 16372 CrossRef CAS PubMed.
- For radical–radical coupling: (a) W. Liu, Y. Li, K. Liu and Z. Li, J. Am. Chem. Soc., 2011, 133, 10756 CrossRef CAS PubMed; (b) Z. Liu, J. Zhang, S. Chen, E. Shi, Y. Xu and X. Wan, Angew. Chem., Int. Ed., 2012, 51, 3231 CrossRef CAS PubMed.
- M. Lian, Q. Li, Y. Zhu, G. Yin and A. X. Wu, Tetrahedron, 2012, 68, 9598 CrossRef CAS PubMed.
- T. B. Nguyen, R. Pascal and A.-M. Ali, Org. Lett., 2013, 15, 5238 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, spectral data and copies of spectra. See DOI: 10.1039/c5ra08030d |
| This journal is © The Royal Society of Chemistry 2015 |