
Bipyridine-functionalized amphiphilic block copolymers as support materials for the aerobic oxidation of primary alcohols in aqueous media
Henning Sand and
Ralf Weberskirch*
Faculty of Chemistry and Chemical Biology, TU Dortmund, Otto-Hahn Str. 6, D 44227 Dortmund, Germany. E-mail: Ralf.Weberskirch@tu-dortmund.de
First published on 21st April 2015
Abstract
Amphiphilic block copolymers with 4-methoxy-4′-alkoxybipyridine ligands in the hydrophobic block were synthesized by cationic ring-opening polymerization. The bipyridine moiety was either introduced directly as a 2-oxazoline monomer (P1) or by polymer-analogous coupling to a precursor poly(2-oxazoline) with chloropentyl side chains (PP2–PP7) to prepare the polymer ligands (P2–P7). The polymers were characterized by NMR and SEC measurements to determine polymer composition, molar masses and polydispersities. In water, these polymers form micelles with cmc values ranging from 1.8 to 22 μmol l−1. SAXS and DLS measurements exhibited spherical particles with particle sizes of 8 to 21 nm. Polymers P1–P7 were finally utilized to carry out the aerobic oxidation of primary alcohols, including allylic, benzylic, and aliphatic derivatives at room temperature (T = 20 °C) and ambient air in aqueous media indicating higher activities for P2–P7 compared to P1 as a consequence of the different preparation methods. Moreover, product isolation and catalyst recycling can be easily accomplished by solvent extraction five times without significant loss of activity.
Introduction
The selective oxidation of primary and secondary alcohols into the corresponding aldehydes or ketones is a key transformation in modern organic synthesis.1–3 While in the past this reaction was carried out by the usage of stoichiometric amounts of inorganic oxidants such as KMnO4,4 CrO3,5 or dichromate,6 it is not surprising that there has been much research devoted to the replacement of these often hazardous or toxic agents by more environmentally friendly chemistry and more active catalysts.7–9 2,2,6,6-Tetramethyl-piperidine-1-oxyl (TEMPO) radicals such as 4-methoxy-TEMPO (1) have been used extensively as a metal-free approach for the selective oxidation of alcohols to aldehydes and ketones.10 The strategy requires only catalytic amounts (typically, <10 mol%) of TEMPO and stoichiometric amounts of a co-oxidants, e.g., sodium hypochlorite,11,12 sodium chlorite,13 or oxone.14Several TEMPO catalyst systems have been described that address also the problem of catalyst recovery and simplified product isolation. Some examples of support materials for TEMPO include silica,15 or multiwall CNTs,16 and more recently various nanoparticles based on Fe3O4,17 Merrifield resins,18 magnetic polystyrene particles,19 and TEMPO polymer-grafted silica nanoparticles have been successfully employed.20 The nanoparticle approach allows very often simplified catalyst isolation and product separation while achieving good activities and thus represents an interesting alternative to homogeneous, soluble polymer supports based on poly(norbornene)21 or PEG.22
On the other side some highly efficient catalysts for the aerobic oxidation of alcohols have been developed recently using either more expensive transition metal complexes based on water-soluble complexes of palladium(II) with phenanthroline-based ligands,23,24 water-soluble platinum(II) tetrasulfophthalocyanine catalysts,25 water-soluble diruthenium complex Ru2(m-OAc)3(m-CO3)26 or RuCl2(PPh3)3/TEMPO catalysts27 and various Rh catalysts.28 In 1984, Semmelhack and coworker reported the first aerobic oxidation of activated primary alcohols with a CuCl/TEMPO catalyst system which was able to oxidize allylic and benzylic alcohols efficiently.29 Despite many advances over the past decades, even the most versatile and active catalyst systems such as the CuCl/1,10-phenanthroline catalyst with dialkylazodicarboxylates as redox-active co catalyst required the use of pure O2 as the oxidant and fluorobenzene as the solvent to obtained optimal results in a wide range of diversely functionalized primary and secondary allylic, benzylic, and aliphatic alcohols.30
More recently, Stahl and coworker described a (bpy)Cu(I)/nitroxyl co-catalyst system that enables the efficient and selective aerobic oxidation of a broad range of primary alcohols, including allylic, benzylic, and aliphatic derivatives, to the corresponding aldehydes.31,32 While many TEMPO-supported catalyst systems have been described in the literature15–21 there have been only few reports on the immobilization of bipyridine ligands to a homogenous polymer support for catalysis application.33–35 Another interesting feature for the development of environmentally friendly catalysts is a support material that is compatible with water and at the same time allows the conversion of hydrophobic substrates. Especially micelles based on block copolymers have been proven particularly successful in the past to provide hydrophobic cavities for the efficient catalytic transformation of hydrophobic substrates in aqueous media.36–38
Herein, we report the first synthesis of well-defined amphiphilic block copolymers modified with bipyridine ligands and their application in the Cu(I)/TEMPO based aerobic oxidation of alcohols in aqueous media.
Experimental
Materials
All chemicals were purchased commercially and used, unless otherwise noted, without further purification. Water-free dichloromethane and dimethylformamide were purchased from Acros Organics and dried over Al2O3 using a M. Braun GmbH MB SPS 800. Other water-free solvents were dried under standard procedures (acetonitrile and chlorobenzene, CaCl2; isopropanol, Mg) and stored over molecular sieve 3 Å and argon atmosphere.Measurements
1H (500.13 MHz) and 13C NMR (100.63 MHz) spectra were recorded on a Bruker DRX 500 spectrometer. GC-EI-HRMS measurements were performed on a Thermo Electro at 160 °C and 70 eV, as reference was used perfluorokerosene. Gel permeation chromatography (SEC) was carried out on a DMF (5 g l−1 LiBr) based SEC at 60 °C with PSS GRAM analytical 1000 Å and 30 Å columns equipped with a Knauer RI detector Smartline 2300 using linear polystyrene standards for the poly(2-oxazoline). Gas chromatography analyses were performed on a Fisons 9000 equipped with a flame ionization detector FID/1177, capillary column CP-Sil 8, length 30 m, with helium as mobile phase and analysed with the program EZChrom Elite. Dynamic light scattering was carried out on a Malvern Zetasizer Nano-Z5 with a HeNe-Laser (λ = 632 nm). Fluorescence spectroscopy was recorded on a Perkin-Elmer LS 55 using a wavelength of λ = 334 nm and a 0.01 mM solution of pyrene in methanol. The analyses were carried out with the program FL WinLab. SAXS measurements were carried out with an Anton Paar SAXSess mc2 Station using 5 mM polymer solutions and a copper anode. Imaging plates were used for detection and for analysis the software Optiquant and SAXSQuant 1D/2D.Monomer synthesis
1H-NMR (500.13 MHz, CDCl3). δ = 0.86 ppm (m, CH3), 1.30 (m, CH3CH2CH2CH2CH2CH2), 1.61 (quin, J = 7.4 Hz, CH3CH2), 2.25 (t, J = 7.7 Hz, CCH2), 3.80 (t, J = 9.4 Hz, CH2N), 4.20 (t, J = 9.5 Hz, CH2O).
13C-NMR (100.63 MHz, CDCl3). δ = 14.0 ppm (CH3), 22.6 (CH3CH2), 25.9 (CH2CH2CO), 27.9 (CH2CH2CH2CH3), 28.9 (CH2CN), 29.2 (CH2CH2CH2CN), 31.6 (CH2CH2CH3), 54.3 (CH2N), 67.1 (CH2O), 168.6 (CO).
ESI-MS. Mcalculated = 169.1467; Mmeasured = 170.1541 [M + H]+.
1H-NMR (500.13 MHz, CDCl3). δ = 1.47 ppm (m, CH2CH2CH2Cl), 1.63 (quin, J = 7.6 Hz, CH2CH2C(O)N), 1.76 (quin, J = 7.2 Hz, CH2CH2O), 2.25 (t, J = 7.5 Hz, CH2C(O)N), 3.5 (t, J = 6.7 Hz, NCH2CH2O), 3.78 (t, 3J = 9.7 Hz, CH2Cl), 4.19 (t, J = 9.5 Hz, NCH2CH2O).
13C-NMR (100.63 MHz, CDCl3). δ = 25.1 ppm (CH2CH2C(O)N), 26.3 (CH2CH2CH2C(O)N), 27.6 (CH2CH2CH2Cl), 32.1 (CH2C(O)N), 44.7 (CH2N), 54.3 (CH2CH2CH2Cl), 67.1 (NCH2CH2O), 168.1 (C(N)O).
ESI-MS. Mcalculated = 175.0764; Mmeasured = 176.0838 [M + H]+.
1H-NMR (500.13 MHz, CDCl3). δ = 3.91 ppm (s, CH3O), 6.53 (dd, J = 7.2 Hz, CHCHCOCH3), 6.88 (dd, J = 5.8 Hz, CHCHCOH), 7.11 (s, br, CCHCOH), 7.39 (s, CCHCOCH3), 7.71 (d, J = 7 Hz, CHCHCOH), 8.42 (d, J = 5.8 Hz, CHCHCOCH3).
13C-NMR (100.63 MHz, CDCl3). δ = 55.5 ppm (CH3O), 106.1 (2 × CCHCOR), 111.2 (2 × CHCHCOR), 150.2 (2 × CHCHCOR), 167.0 (2 × NCCH, 2 × COR).
ESI-MS. Mcalculated = 202.0742; Mmeasured = 203.0816 [M + H]+.
1H-NMR (500.13 MHz, CDCl3). δ = 1.56 pp (m, CH2CH2CH2O), 1.72 (m, CH2CH2C(O)N), 1.85 (m, CH2CH2O), 2.32 (t, J = 7.5 Hz, CH2C(O)N), 3.82 (t, J = 9.4 Hz, NCH2CH2O), 3.95 (s, CH3O), 4.13 (t, J = 6.4 Hz, CH2O), 4.22 (t, J = 9.5 Hz, NCH2CH2O), 6.83 (ddd, J = 8/5.5/2.5 Hz, 2 × CHCHC), 7.95 (dd, J = 8.7/2.4 Hz, 2 × CCHC), 8.46 (dd, J = 5.5/3.3 Hz, 2 × NCH).
13C-NMR (100.63 MHz, CDCl3). δ = 25.5 ppm (CH2CH2CN), 25.6 (CH2CH2CH2CN), 27.8 (CH2CH2CH2O), 28.6 (CH2CN), 54.3 (CH2N), 55.3 (CH3O), 67.2 (NCH2CH2O), 67.7 (CH2CH2CH2O), 106.0 (CCHCOCH3), 106.7 (CCHCOCH2), 111.0 (CHCHCOCH3), 111.3 (CHCHCOCH2), 150.0 (2 × NCH), 157.7 (2 × CHNC), 166.0 (CHCOCH2), 166.6 (CHCOCH3), 168.3 (C(N)O).
ESI-MS. Mcalculated = 341.1739; Mmeasured = 342.1818 [M + H]+.
Polymer synthesis
All polymerizations were carried out in Schlenk tubes under inert atmosphere using freshly distilled and dried solvents. A typical procedure was as follows:To 2-methyl-2-oxazoline (495.05 μl, 5.88 mmol, 22 eq.) in 7 ml acetonitrile was given methyltriflate (30.22 μl, 267.05 μmol, 1 eq.) at 0 °C. After stirring the mixture for 3 h at 120 °C, M2 (273.52 mg, 801.15 μmol, 3 eq.), 3 ml chlorobenzene and 2-heptyl-2-oxazoline (188.34 μl, 1.07 mmol, 4 eq.) were added. The mixture was stirred for 24 h at 100 °C and terminated at rt by addition of piperidine (79.27 μl, 801.15 μmol, 3 eq.) for 12 h. After removal of the solvent, the residue was dissolved in 10 ml of chloroform and stirred with K2CO3 for 3 h. After filtration, P1 was purified by precipitation in ice cold diethyl ether and dried under reduced pressure.
1H-NMR (500.13 MHz, CDCl3). δ = 0.85 ppm (s, NC(O)C6H12CH3), 1.26 (s, NC(O)CH2CH2C4H8CH3), 1.39–1.84 (NC(O)CH2C3H6CH2Cl/NC(O)CH2CH2C5H11/NCH2CH2CH2CH2CH2), 2.00–2.20 (NC(O)CH3), 2.31 (s, NC(O)CH2C6H13/NC(O)CH2C4H8Cl), 2.90–3.05 (NCH3), 3.45 (s, NCH2CH2N/CH2Cl).
To PP2 (1 eq. related to Cl-functionality) in dimethylformamide was given L2 (1.2 eq.) and K2CO3 (1.2 eq.). After stirring the mixture for 36 h at 130 °C the solvent was removed. The residue was dissolved in dichloromethane, filtrated and the solvent was removed. The polymer was purified by precipitation in ice cold diethyl ether and dialyzed against ethanol (MWCO 1000) for 24 h. After removal of the solvent, polymer P2 was again precipitated in ice cold diethyl ether and obtained as a white powder after drying.
P1/P2: 1H-NMR (500.13 MHz, CDCl3). δ = 0.85 ppm (s, NC(O)C6H12CH3), 1.26 (s, NC(O)CH2CH2C4H8CH3), 1.45–1.90 (NC(O)CH2C3H6CH2OBiPy/NC(O)CH2CH2C5H11/NCH2CH2CH2CH2CH2), 2.00–2.20 (NC(O)CH3), 2.31 (s, NC(O)CH2C6H13/NC(O)CH2C4H8OBiPy), 2.90–3.05 (NCH3), 3.45 (s, NCH2CH2N), 3.92 (s, CH3O), 4.10 (s, CH2OBiPy), 6.81 (s, 2 × CHCHC), 7.93 (s, 2 × CCHC), 8.43 (s, 2 × NCH).
To isolate the product, the solution was extracted with diethyl ether (4 × 20 ml). The organic layers were dried with MgSO4 and removed under reduced pressure. The product was purified via column chromatography (cyclohexane/ethyl acetate 5/1) and yielded with 94%. The catalyst solution in water could be used for further runs by just adding N-methylimidazole and TEMPO again. For kinetics, 50 μl was taken out of the reaction solution and was frozen in liquid nitrogen. 30 μl of standard solution (200 μl n-undecane in 2 ml cyclohexane) and 500 μl diethyl ether were added. After extraction, MgSO4 was added into the vial, the remaining solution was purged through a mirco-cloumn and the vial was washed with 500 μl diethyl ether again. The conversion was determined by gas chromatography.
Results and discussion
Synthesis of amphiphilic, bipyridine functionalized block copolymers
The cationic ring-opening polymerization of 2-oxazolines provides a versatile monomer system for the synthesis of amphiphilic block copolymers with different architecture, composition and fuctionalization.40,41 Two different approaches can be utilized to incorporate the bipyridine units into polyoxazolines, either (i) directly via suitable bipyridine-containing monomers42 or (ii) in a polymer-analogous fashion by coupling of the bipyridine moiety to a precursor polymer as has been demonstrated for other ligands before.43 While the first approach is clearly more elegant, the post-analogous modification of a precursor polymer has been proven to be very useful when the ligand may interfere with the cationic polymerization procedure.43 Therefore, we decided to develop a monomer synthesis route that allows the examination of both approaches of bipyridine introduction into the polymer. The synthesis of the monomers M1 and M2 is shown in Scheme 1. As a ligand we chose the 4-methoxy-4′-alkoxybipyridine system which has recently shown to be more active in the aerobic oxidation of primary alcohols than the bipyridine ligands.44 | ||
| Scheme 1 Synthesis of the 2-oxazoline monomers M1 and M2. | ||
The synthesis of (2-(5-chloropentyl)-2-oxazoline) monomer M1 was carried out according to a literature procedure45 starting from ε-caprolactone and its ring-opening reaction with ethanolamine. Overall yield for the three step synthesis was 66%. The bipyridine-containing monomer M2 was prepared by reacting M1 with the deprotonated bipyridine derivative L2 in a Williamson ether synthesis to give M2 in 94% yield.
The structure and composition of M2 were confirmed by 1H and 13C NMR spectroscopy and mass spectrometry. Characteristic are the two methylene groups of the oxazoline ring at 3.82 (–N–CH2–) and 4.22 ppm (–O–CH2–) and the signals at 6.83, 7.95 and 8.46 ppm can be clearly assigned to the protons of the bipyridine unit.
Two approaches were pursued to prepare catalytically active polymer scaffolds (see Scheme 2). In the first approach (Route A), we prepared the amphiphilic block copolymer by consecutive cationic polymerization of 2-methyl-2-oxazoline to form the hydrophilic block and a mixture of M2/HepOx to form the hydrophobic block. The polymerization was terminated with piperidine. The polymer structure of P1 was analyzed by 1H NMR spectroscopy (see Fig. 1) that confirmed the successful introduction of M2 into the hydrophobic block with the signals 12–14 corresponding to the bipyridine unit between 6.9 to 8.5 ppm.
 | ||
| Scheme 2 Synthesis of the bipyridine-containing poly(2-oxazoline)s P1–P7. A: direct introduction of the bipyridine unit by polymerization of M2. B: post-analogous coupling of the bipyridine moiety to the polymer precursor PP2–PP7 in a Williamson ether synthesis (indirect method). | ||
 | ||
| Fig. 1 1H-NMR (CDCl3, 500.13 MHz) of the bipyridine-containing copolymer P1. | ||
Moreover, 1H NMR spectroscopy was used to analyze copolymer composition and molar mass by end group analysis (see Table 1). Size exclusion chromatography of P1, however, resulted in a bimodal curve and a polydispersity index of ![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) w/n = 1.34. The bimodal shape of the SEC curve for P1 can be explained by a partial termination of the cationic chain end by the bipyridine unit during polymerization (see Fig. 2). As a consequence, part of the bipyridine moieties may not be available anymore for the formation of the active catalyst in P1.
w/n = 1.34. The bimodal shape of the SEC curve for P1 can be explained by a partial termination of the cationic chain end by the bipyridine unit during polymerization (see Fig. 2). As a consequence, part of the bipyridine moieties may not be available anymore for the formation of the active catalyst in P1.
| Polymer | Polymer compositiona | MN NMRb (g mol−1) | MN SECc (g mol−1) | PDIc | cmcd (×10−6 mol l−1) | dDLSe (nm) | dSAXSf (nm) |
|---|---|---|---|---|---|---|---|
| a Polymer composition as determined by 1H NMR analysis; Me = 2-methyl-2-oxazolin; Hep = 2-heptyl-2-oxazolin; BiPy = 4-methoxy-4-pentoxy-2,2-bipyiridne units.b Determined by 1H NMR endgroup analysis in CDCl3.c Determined by SEC analysis in DMF/5 g l−1 LiBr with linear PS standards.d With pyrene (0.01 mM in MeOH).e By DLS measurements in water (0.5–1 mM) at room temperature.f By SAXS measurements in water (1–5 mM) at room temperature. | |||||||
| P1 | Me25Hep3BiPy2 | 3450 | 3490 | 1.34 | 7.9 | 11.01 | n. d. |
| P2 | Me26Hep3BiPy2 | 3530 | 4010 | 1.09 | 7.9 | 8.04 | 11.68 |
| P3 | Me37Hep2BiPy2 | 4310 | 4440 | 1.18 | 22 | 10.74 | 13.12 |
| P4 | Me40Hep5BiPy4 | 5770 | 4660 | 1.15 | 1.8 | 12.48 | 13.38 |
| P5 | Me20BiPy3 | 2850 | 3030 | 1.09 | 18 | 11.86 | 13.90 |
| P6 | Me38BiPy5 | 5080 | 3230 | 1.20 | 20 | 16.11 | n. d. |
| P7 | Me50BiPy10 | 7839 | 4940 | 1.18 | 3.1 | 21.16 | 19.08 |
 | ||
| Fig. 2 SEC curve for P1 (direct polymerization) and P2 (polymer-analogous introduction of the bipyridine ligand). | ||
Therefore, all other polymers P2–P7 were prepared in a two-step approach, by synthesizing first a polymer precursors (PP2–PP7) with M1 in the hydrophobic block, that were then reacted in a polymer-analogous fashion with the bipyridine moiety L2 to give the final polymer ligands P2–P7 (see Scheme 2, Route B). By using this methodology we prepared two sets of polymer ligands that contained either a mixture of 2-heptyl-2-oxazoline and the 4-methoxy-4′-pentoxy-2,2′-bipyridine ligand in the side chain of the hydrophobic block (P2–P4) or only the bipyridine moiety as the hydrophobic unit (P5–P7). Narrow PDI values of 1.09–1.20 suggest a living polymerization process that was further validated by mono-modal SEC curves for all polymer P2–P7 as can be seen for P2 in Fig. 2. All polymers P2–P7 were characterized by 1H NMR spectroscopy to determine their composition and molecular weight by endgroup analysis (see Table 1).
Characterization of micellar aggregates
The study of aggregate formation was guided by two considerations. First, we wanted to make sure that micellar aggregates were formed at the polymer concentrations used in the catalytic experiments. In addition, we were interested in finding out if there is any correlation between copolymer composition, aggregate size and catalytic activity. Critical micelle concentration (cmc) was determined for each copolymer P1–P7 by pyrene solubilization experiments resulting in cmc values of 3.1 to 20 μmol l−1 which is in a typical range for amphiphilic poly(oxazoline) block copolymers.36,43 Aggregate size and shape of the polymers P1–P7 formed in water was further analyzed by DLS and SAXS measurements at a similar polymer concentration of 5 mM polymer solutions that were also used in the subsequent catalysis experiments. The results are summarized in Table 1, column 6–8. The hydrodynamic radii of the polymer micelles were in the range of 8 to 21 nm which is typical for micellar aggregates based on poly(2-oxazoline)s with a similar molar mass and block copolymer composition and a rather bulky, hydrophobic side chain.36,43 Moreover it was noticed that polymers (P5–P7) that contained only bipyridine units in the hydrophobic block formed larger aggregates with diameter of 12 to 21 nm compared to polymers (P2–P4) where the hydrophobic block was composed of a mixture of heptyl side chains and bipyridine units with 8 to 12.5 nm. SAXS measurements confirmed the size of the micellar aggregates and showed that all particles displayed a spherical shape (data not shown here).Application of the polymer micelles in the aerobic oxidation of primary alcohols
The homogeneous, aerobic oxidation of primary alcohols catalyzed by a (bpy)Cu(I)/nitroxyl co-catalyst system has witnessed great interest recently due to their high activity and selectivity for a broad range of primary alcohols.31,32 Although homogeneous catalysts have many advantages, catalyst immobilization is a well known methodology to allow efficient catalyst separation and to obtain metal-free products.46–51 Moreover, with the increasing interest in Green Chemistry Processing the replacement of expensive, toxic and flammable organic solvents by water as the preferred solvent is highly desirable due to economically and safety related process engineering reasons.52–55In the first set of experiments we studied the effect of polymer-ligand preparation, P1 (direct polymerization) and P2 (polymer-analogous introduction of the bipyridine ligand) on catalytic activity. Cu(I)Br was chosen as the copper source due to its higher stability in aqueous media compared to [Cu(MeCN)4]OTf which is preferred in organic media. The active catalyst was prepared by dissolution of the polymer-ligand in acetonitrile (5 mM polymer solution) before CuBr was added in stoichiometric amounts with respect to the bipyridine units to the solution. After 1 h stirring, the solvent was removed and the Cu–polymer complex was dissolved in water before N-methylimidazole, TEMPO and benzyl alcohol were added to the mixture. As can be seen from Fig. 3A, the synthetic route of polymer-ligand preparation had a strong effect on catalytic activity. With P1 a nearly quantitative conversion of benzyl alcohol to benzaldehyde was obtained after 7 h whereas with P2 it was possible to obtain this after only 3 h. The results confirm our previous findings from the SEC analysis of P1 and P2. The polymerization of M2 leads to a partial termination of the polymerization reaction in P1 due to the nucleophilic character of the bipyridine units. As a result less ligand is available for the formation of an active complex thus leading to a reduced activity.
 | ||
| Fig. 3 A oxidation of benzyl alcohol (5 mol% Cu(I)Br; 5 mol% ligand, 5 mol% TEMPO, 10 mol% NMI, solvent, at RT, air) with P1 and P2 in water. B: comparison of the conversion of representative benzylic (A1), allylic (A2) and aliphatic alcohol (A3) under micellar catalytic conditions with P6. | ||
In the next set of experiments we studied the conversion of different benzylic, allylic and aliphatic alcohols under micellar catalytic conditions with P6 as the catalyst support. As can be seen from Fig. 3B, benzyl alcohol was completely converted after 180 min, while the conversion of allylic alcohol and aliphatic alcohol was considerable slower with 70% and 10%, respectively. This gradual decrease in reactivity from benzylic and allylic to aliphatic alcohols is in excellent agreement with the results reported in the literature from homogeneous oxidation experiments (Table 2).31,32




Although polymers P2–P7 varied in size (d = 8 to 21 nm) and composition of the hydrophobic polymer block no significant difference in catalytic activity was observed (see Table 3) which again underlines the robustness and flexibility of the polymeric carrier system.
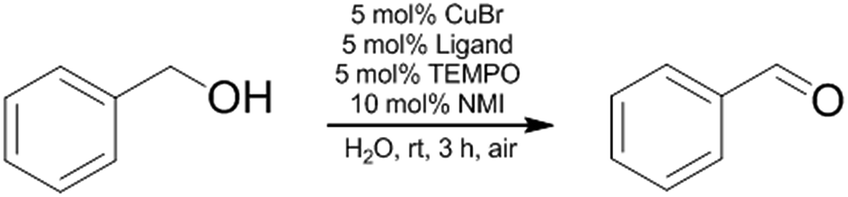
The main advantage of the polymer-supported catalyst system besides working in aqueous media is the possibility of catalyst recycling. Separation of the catalyst and the product is feasible by simple extraction of the aqueous solution. Therefore, we used diethyl ether as extracting solvent to isolate the product(s) while the polymer bound bipyridine Cu catalyst remains in the aqueous phase. Only TEMPO and NMI had to be given to the aqueous polymer phase after every extraction step. In contrast, the use of low molecular weight surfactants with a catalyst that is not bound to the surfactant, leads often to catalyst leaching and recycling is not possible by simple extraction.56–59 As can be seen in Fig. 4, the catalyst system shows no significant decrease in reactivity in 5 cycles.
 | ||
| Fig. 4 Recycling experiments for the oxidation of benzyl alcohol in H2O in the presence of P6. | ||
Conclusion
In summary, we developed a route to well-defined bipyridine-supported amphiphilic block copolymers for the micellar catalytic aerobic oxidation of benzylic, allylic and aliphatic primary alcohols. Successful recycling makes these polymer supports very attractive for the preparation of various aldehydes under environmental benign reaction conditions.Acknowledgements
We are grateful to Paul Hendrik Schummel for performing the pyrene solubilisation experiments as well as the DLS measurements and Dr Mirko Erlkamp for performing the SAXS-measurements (both group of Prof. R. Winter/PC I/TU Dortmund).Notes and references
- S. V. Ley, J. Norman, W. P. Griffith and S. P. Marsden, Synthesis, 1994, 639–666 CrossRef.
- R. A. Sheldon, I. W. C. E. Arends, G. J. Ten Brink and A. Dijksman, Acc. Chem. Res., 2002, 35, 774–781 CrossRef CAS PubMed.
- Y. Ishii, S. Sakaguchi and T. Iwahama, Adv. Synth. Catal., 2001, 343, 393–427 CrossRef CAS.
- F. M. Menger and C. Lee, Tetrahedron Lett., 1981, 22, 1655–1656 CrossRef CAS.
- J. R. Holum, J. Org. Chem., 1961, 26, 4814–4816 CrossRef CAS.
- D. G. Lee and U. A. Spitzer, J. Org. Chem., 1970, 35, 3589–3590 CrossRef CAS.
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC.
- B. L. Conley, M. K. Pennington-Boggio, E. Boz and T. J. Williams, Chem. Rev., 2010, 110, 2294–2312 CrossRef CAS PubMed.
- R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2010, 14, 245–251 CrossRef CAS.
- P. L. Anelli, C. Biffi, F. Montanari and S. Quici, J. Org. Chem., 1987, 52, 2559–2562 CrossRef.
- A. E. J. de Nooy, A. C. Besemer and H. V. Bekkum, Synthesis, 1996, 10, 1153–1174 CrossRef PubMed.
- T. Miyazawa, T. Endo, S. Shiihashi and M. Okawara, J. Org. Chem., 1985, 50, 1332–1334 CrossRef CAS.
- M. Zhao, J. Li, E. Mano, Z. Song, D. M. Tschaen, E. J. J. Grabowski and P. J. Reider, J. Org. Chem., 1999, 64, 2564–2566 CrossRef CAS.
- C. Bolm, A. S. Magnus and J. P. Hildebrand, Org. Lett., 2000, 2, 1173–1175 CrossRef CAS.
- C. Bolm and T. Fey, Chem. Commun., 1999, 1795–1796 RSC.
- Y. Wang, X. Y. Song, S. H. Shao, H. M. Zhong and F. Lin, RCS Adv., 2013, 2, 7693–7698 Search PubMed.
- A. K. Tukcer-Schwartz and R. L. Gareell, Chem.–Eur. J., 2010, 16, 12718–12726 CrossRef PubMed.
- A. Gheorghe, A. Matsuno and O. Reiser, Adv. Synth. Catal., 2006, 348, 1016–1020 CrossRef CAS PubMed.
- Z. Zheng, J. L. Wang, M. Zhang, L. X. Xu and J. B. Li, ChemCatChem, 2013, 5, 307–312 CrossRef CAS PubMed.
- K. Saito, K. Hirose, T. Okayasu, H. Nishide and M. T. W. Hearn, RSC Adv., 2013, 3, 9752–9756 RSC.
- G. Tanyeli and A. Gümüş, Tetrahedron Lett., 2003, 44, 1639–1642 CrossRef.
- P. Ferreira, E. Phillips, D. Rippon, S. Chi Tsang and W. Hayes, J. Org. Chem., 2004, 69, 6851–6859 CrossRef CAS PubMed.
- M. Verhoef, J. Peters and H. van Bekkum, Stud. Surf. Sci. Catal., 1999, 125, 465–472 CrossRef CAS.
- G. J. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Adv. Synth. Catal., 2002, 344, 355–369 CrossRef CAS.
- L. Tonucci, M. Nicastro, N. d'Alessandro, M. Bressan, P. D'Ambrosio and A. Morvillo, Green Chem., 2009, 11, 816–820 RSC.
- N. Komiya, T. Nakae, H. Sato and T. Naota, Chem. Commun., 2006, 46, 4829–4831 RSC.
- A. Dijksman, A. Marino-González, A. Mairata i Payeras, I. W. C. E. Arends and R. A. Sheldon, J. Am. Chem. Soc., 2001, 123, 6826–6833 CrossRef CAS PubMed.
- T. Zweifel, J. V. Naubron and H. Grützmacher, Angew. Chem., Int. Ed., 2009, 48, 559–563 CrossRef CAS PubMed.
- M. F. Semmelhack, C. R. Schmid, D. A. Cortes and C. S. Chou, J. Am. Chem. Soc., 1984, 106, 3374–3376 CrossRef CAS.
- I. E. Mark, P. R. Giles, M. Tsukazaki, S. M. Brown and C. J. Urch, Science, 1996, 274, 2044–2046 CrossRef.
- J. M. Hoover and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 16901–16910 CrossRef CAS PubMed.
- J. E. Steves and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 15742–15745 CrossRef CAS PubMed.
- T. Kotre, O. Nuyken and R. Webeskirch, Macromol. Chem. Phys., 2004, 205, 1187–1195 CrossRef CAS PubMed.
- C. Wan, Y. Chung and P. H. Toy, J. Comb. Chem., 2007, 9, 115–120 CrossRef PubMed.
- J. V. Nguyen and C. W. Jones, Macromolecules, 2004, 37, 1190–1203 CrossRef CAS.
- D. Schönfelder, K. Fischer, M. Schmidt, O. Nuyken and R. Weberskirch, Macromolecules, 2005, 38, 254–262 CrossRef.
- M. Bortenschlager, N. Schöllhorn, A. Wittmann and R. Weberskirch, Chem.–Eur. J., 2007, 13, 520–527 CrossRef CAS PubMed.
- A. Lu, P. Cotanda, J. P. Patterson, D. A. Longbottom and R. K. O'Reilly, Chem. Commun., 2012, 48, 9699–9701 RSC.
- J. Kim and M. H. Litt, J. Polym. Sci., 1989, 27, 2711–2722 CrossRef CAS PubMed.
- S. Kobayashi and H. Uyama, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 192–209 CrossRef CAS PubMed.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978–7994 CrossRef CAS PubMed.
- T. Kotre, O. Nuyken and R. Weberskirch, Macromol. Rapid Commun., 2002, 23, 871–876 CrossRef CAS.
- B. M. Rossbach, K. Leopold and R. Weberskirch, Angew. Chem., Int. Ed., 2006, 45, 1309–1312 CrossRef CAS PubMed.
- D. Könning, W. Hiller and M. Christmann, Org. Lett., 2012, 14, 5258–5261 CrossRef PubMed.
- A. Levy and M. Litt, J. Polym. Sci., Part A: Gen. Pap., 1968, 16, 1883–1891 CrossRef PubMed.
- C. A. McNamara, M. J. Dixon and M. Bradley, Chem. Rev., 2002, 102, 3275–3300 CrossRef CAS PubMed.
- N. E. Leadbeater and M. Marco, Chem. Rev., 2002, 102, 3217–3274 CrossRef CAS PubMed.
- H. P. Dijkstra, G. P. M. Van Klink and G. van Koten, Acc. Chem. Res., 2002, 35, 798–810 CrossRef CAS PubMed.
- T. J. Dickerson, N. N. Reed and K. D. Janda, Chem. Rev., 2002, 102, 3325–3344 CrossRef CAS PubMed.
- N. Sahiner, Prog. Polym. Sci., 2013, 38, 1329–1356 CrossRef CAS PubMed.
- D. E. Bergbreiter, J. Tian and C. Hongfa, Chem. Rev., 2009, 109, 530–582 CrossRef CAS PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed.
- P. Tundo, P. Anastas, D. S. Black, J. Breen, T. Collins, S. Memoli, J. Miyamoto, M. Polyakoff and W. Tumas, Pure Appl. Chem., 2000, 72, 1207–1228 CAS.
- B. Cornils and W. A. Herrmann, Aqueous Phase Organometallic Catalysis – Concept and Application, Wiley–VCH, Weinheim, 1998 Search PubMed.
- M. B. Gawande, V. D. B. Bonifacio, R. Luque, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 5522–5551 RSC.
- T. Dwars, E. Paetzold and G. Oehme, Angew. Chem., Int. Ed., 2005, 44, 7174–7199 CrossRef CAS PubMed.
- G. Oehme, I. Grassert, E. Paetzold, H. Furhmann, T. Dwars, U. Schmidt and I. Iovel, Kinet. Catal., 2003, 44, 766–777 CrossRef CAS.
- B. H. Lipshutz, M. Hageman, J. C. Fennewald, R. Linstadt, E. Slack and K. Voigtritter, Chem. Commun., 2014, 50, 11378–11381 RSC.
- T. Dwars, J. Haberland, I. Grassert, G. Oehme and U. Kragl, J. Mol. Catal. A: Chem., 2001, 168, 81–86 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |