
One-pot synthesis of poly(l-lactide)-b-poly(methyl methacrylate) block copolymers†
Abstract
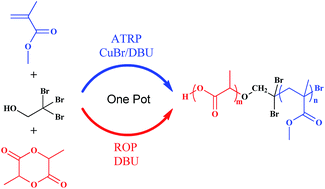
By using 1,8-diazabicyclo-[5.4.0]undec-7-ene (DBU) as both the atom transfer radical polymerization (ATRP) ligand and ring-opening polymerization (ROP) catalyst, we have combined copper-catalyzed ATRP and organo-catalyzed ROP in one-pot, and successfully synthesized poly(L-lactide)-b-poly(methyl methacrylate) (PLA-b-PMMA) diblock copolymers. The structure of the block copolymers is confirmed by nuclear magnetic resonance (NMR), size exclusion chromatography (SEC) and differential scanning calorimetry (DSC).
 Please wait while we load your content...
Please wait while we load your content...

