
Hydroxyl radical-induced etching of glutathione-capped gold nanoparticles to oligomeric AuI–thiolate complexes†
Tzu-Heng Chena,
Chih-Chun Nieha,
Ya-Chen Shiha,
Chen-Yi Kea and
Wei-Lung Tseng*ab
aDepartment of Chemistry, National Sun Yat-sen University, Taiwan. E-mail: tsengwl@mail.nsysu.edu.tw
bSchool of Pharmacy, College of Pharmacy, Kaohsiung Medical University, Taiwan
First published on 12th May 2015
Abstract
Thiol-induced core etching of gold nanoparticles is a general method for the production of gold nanoclusters (AuNCs) of various sizes. This paper is the first report on the efficient reaction of glutathione-capped gold nanoparticles (GSH-AuNPs) with hydroxyl radicals to produce oligomeric AuI–thiolate complexes at ambient temperature. Also, hydroxyl radicals can etch commercially available gold nanoparticles (100 nm); this strategy can be applied for the removal of gold from scrap electronics. Additionally, proteins can trigger the aggregation of oligomeric AuI–thiolate complexes under neutral conditions resulting in the formation of fluorescent AuNCs. For example, the reaction of trypsin, lysozyme, and glucose oxidase with oligomeric AuI–thiolate complexes produces Au5, Au8, and Au13 clusters with emission maxima at 415, 460, and 535 nm, respectively. Interestingly, trypsin- and glucose oxidase-stabilized AuNCs could sense GSH and glucose via GSH-induced etching of AuNCs and H2O2-mediated oxidation of AuNCs, respectively.
1. Introduction
Gold nanoclusters (AuNCs), which consist of a couple of atoms up to a few hundred atoms, have received significant interest over the past decade because of their good optical, electrical, and catalytic properties.1 Because the sizes of AuNCs range between those of isolated gold atoms and nanoparticles, their discrete energy levels result in molecule-like properties such as size-dependent fluorescence. For example, poly(amidoamine) dendrimer-encapsulated Au5, Au8, Au13, Au25, and Au31 clusters emit UV, blue, green, red, and near-IR light, respectively.2 Compared to organic fluorophores, AuNCs provide the distinct advantages of a one-pot synthesis, large Stokes shift, long fluorescence lifetime, good photostability, and low toxicity. Because of their intrinsic fluorescence, AuNCs are emerging as optical probes for use in the fields of sensors, molecular imaging, flow cytometry, and bioconjugation.3,4Numerous synthetic methods have been developed to produce different sized AuNCs with fluorescent emission ranging from the UV to near-IR regions. Generally, the formation of AuNCs can be achieved by reducing gold ions to their zero-valent state in the presence of thiol-terminated small molecules and a cavity-containing template. Choosing a suitable capping ligand and template is of immense importance to the production of highly stable, fluorescent AuNCs with specific emission wavelengths. For example, near-IR-emitting AuNCs have been prepared via NaBH4 reduction of a gold precursor in the presence of lipoic acid5,6 or glutathione,7 while blue-emitting AuNCs have been synthesized by reducing a mixture of gold precursor and 3-mercaptopropionic acid with NaBH4.8 Hydroxyl-terminated poly(amidoamine) dendrimer and bovine serum albumin were utilized as templates for preparing blue-emitting Au8 and red-emitting Au25 clusters, respectively.9,10 Alternatively, AuNCs can be produced by ligand-induced removal of gold atoms from the surface of nanometer-sized gold particles (>2 nm). The mechanism of ligand-induced etching of gold particles involves electron injection from the ligand to the gold particles.11–13 The type of ligand is one of the most critical factors in determining the size and quantum yield of AuNCs. Long-chain thiols, such as 11-mercatoundecanoic acid,14 dihydrolipoic acid,15 11-mercapto-3,6,9-trioxaundecyl-α-D-mannopyranoside,16 and glutathione,17 are the most commonly used ligands for the top-down approach. However, thiol-containing ligands suffer from low water solubility, strong odors, and air oxidation. Moreover, the top-down approach requires an additional purification process to remove the excess thiols.
The Fenton reaction involves the production of strongly oxidizing hydroxyl radicals via FeII-mediated reduction of hydrogen peroxide.18,19 The formed hydroxyl radicals, which are one of the most powerful oxidizing species, are effective for the degradation of organic pollutants in soils and waters.20–22 Also, the Fenton reaction has been demonstrated to be capable of removing metastable surface gold atoms from a Au surface23,24 and preparing graphene quantum dots from graphene oxide.25 These results suggest that the Fenton reaction could be used to etch gold-based nanomaterials in the absence of thiol-containing ligands.
Herein, we disclose that the hydroxyl radicals in Fenton's reagent quickly etch glutathione (GSH)-capped gold nanoparticles (GSH-AuNPs) to form oligomeric AuI–thiolate complexes. Hydroxyl radical-induced dissolution of GSH-AuNPs was applied to recycle gold from scrap electronics. Also, the addition of trypsin, lysozyme, and glucose oxidase to the oligomeric AuI–thiolate complexes directly produced Au5–, Au8–, and Au13–thiolate clusters, respectively. Trypsin- and glucose oxidase-stabilized Au8 and Au13 clusters were utilized to sense GSH and glucose via thiol-induced core-etching of Au8 clusters and H2O2-mediated oxidation of Au13 clusters, respectively. Scheme 1 illustrates an overview of the disclosed methods.
 | ||
| Scheme 1 Schematic illustration for the process of the synthesis of GSH-AuNPs, hydroxyl radical-triggered etching of GSH-AuNPs, the formation of oligomeric AuI–thiolate complexes, the production of protein-stabilized AuNCs, and the use of protein-stabilized AuNCs as a sensor. | ||
2. Materials and methods
Chemicals
Hydrogen tetrachloroaurate(III) dehydrate was purchased from Alfa-Aesar (Ward Hill, MD, USA). Bovine serum albumin (from bovine serum), NaOH, HCl, NaCl, Na2HPO4, NaH2PO4, H3PO4, Na3PO4, KI, FeCl2, NaBH4, AgNO3, CTAB, GSH, quinine sulfide, ascorbic acid, glucose, glucose oxidase (from Aspergillus niger, 117![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 U g−1), choline oxidase (from Alcaligenes species, 10 U mg−1), α-cyano-4-hydroxy-cinnamic acid, tris(hydroxymethyl)aminomethane (TRIS), and ammonium persulfate were ordered from Sigma-Aldrich (St. Louis, MO, USA). A solution of H2O2 (30%) was obtained from Showa (Tokyo, Japan). Lysozyme (from chicken egg white) and trypsin (from bovine pancreas) were obtained from MP Biomedicals (Irvine, CA). Premixed 30% acrylamide/bisacrylamide solution (29:1), sodium salt of N,N,N′,N′-tetramethylethylenediamine (TEMED), bromophenol blue, and Coomassie brilliant blue R-250 were obtained from Bio-Rad. Milli-Q ultrapure water (Milli-Pore, Hamburg, Germany) was used in all of the experiments.
200 U g−1), choline oxidase (from Alcaligenes species, 10 U mg−1), α-cyano-4-hydroxy-cinnamic acid, tris(hydroxymethyl)aminomethane (TRIS), and ammonium persulfate were ordered from Sigma-Aldrich (St. Louis, MO, USA). A solution of H2O2 (30%) was obtained from Showa (Tokyo, Japan). Lysozyme (from chicken egg white) and trypsin (from bovine pancreas) were obtained from MP Biomedicals (Irvine, CA). Premixed 30% acrylamide/bisacrylamide solution (29:1), sodium salt of N,N,N′,N′-tetramethylethylenediamine (TEMED), bromophenol blue, and Coomassie brilliant blue R-250 were obtained from Bio-Rad. Milli-Q ultrapure water (Milli-Pore, Hamburg, Germany) was used in all of the experiments.
Synthesis of GSH-AuNPs and oligomeric AuI–thiolate complexes
An aqueous solution of HAuCl4 (25 mM, 2 mL) was mixed with GSH (1 mM, 16 mL) under gentle stirring at ambient temperature for 30 min. Subsequently, freshly prepared NaBH4 (10 mg mL−1, 2 mL) was rapidly added to the mixture. The obtained solution was gently stirred at ambient temperature for 12 h. To remove excess GSH and HAuCl4, a solution of GSH-AuNPs was centrifuged at 175000 rpm (34230g) for 10 min. To obtain oligomeric AuI–thiolate complexes, the as-prepared GSH-AuNPs reacted with Fenton's solution (5 mM FeII and 100 mM H2O2.) at ambient temperature for 0–3 h.
Synthesis of gold nanorods
A seed solution was prepared by adding 120 μL of ice-cold 1 mM NaBH4 to 2 mL of a solution containing HAuCl4 (0.05 mM, 1 mL) and CTAB (200 mM, 1 mL). The mixture was stirred until a brownish yellow solution was formed. The formed seed solution (12 μL) was incubated with the growth solution consisted of CTAB (0.2 M, 5 mL), AgNO3 (0.4 mM, 0.1 mL), HAuCl4 (0.1 mM, 5 mL), and ascorbic acid (80 mM, 70 μL) at ambient temperature, resulting in the production of gold nanorods.Synthesis of octahedral gold nanocrystals
A seed solution was prepared by adding 450 μL of ice-cold 0.02 M NaBH4 to 10 mL of a solution containing HAuCl4 (0.05 mM, 5 mL) and CTAB (200 mM, 5 mL). The mixture was left to continue under gentle stirring at ambient temperature for 1 h. Two growth solutions were prepared. For the preparation of the first growth solution (denoted as A), 220 μL of 4 mM ascorbic acid was thoroughly mixed with a solution containing CTAB (0.1 M, 20 mL), KI (0.1 mM, 55 μL), and HAuCl4 (10 mM, 0.25 mL). Similarly, the second growth solution (denoted as B) was obtained by mixing 880 μL of 4 mM ascorbic acid with a solution containing CTAB (0.1 M, 20 mL), KI (0.1 mM, 220 μL), and HAuCl4 (10 mM, 1 mL). Next, 0.1 mL of the seed solution was added to A with gentle shaking until the solution color turned light pink. The obtained solution (0.5 mL) was then transferred to B. After 2 h, the mixture was purified via centrifugation (3000 rpm, 10 min).Synthesis of protein-stabilized AuNCs
A solution of 25 mg mL−1 protein (trypsin, bovine serum albumin, lysozyme, glucose oxidase, and choline oxidase) was added to an equal volume of the as-synthesized oligomeric AuI–thiolate complexes. The resulting mixture was incubated in 5 mM phosphate (pH 7.0) at ambient temperature for 8 h. To remove excess protein, a solution of protein-stabilized AuNCs was dialysed with 5 mM phosphate (pH 7.0). The purified protein-stabilized AuNCs were prepared in 400 mM NaCl and 0–5 mM GSH to test their stability.Sensing of GSH and glucose
Stock solutions of GSH and glucose were prepared in 10 mM phosphate buffer at pH 6. A solution of protein-stabilized AuNCs was diluted to 5-fold with 10 mM phosphate (pH 6.0). Different concentrations of GSH (0–10 mM, 250 μL) were incubated with a solution of trypsin-stabilized AuNCs (250 μL) at ambient temperature for 30 min. After centrifugation at 175000 rpm (34230g) for 3 min, the obtained supernatant was transferred into a 1 mL quartz cuvette. Their fluorescence spectra were recorded by operating the fluorescence spectrophotometer at an excitation wavelength of 350 nm. For sensing glucose, glucose oxidase-stabilized AuNCs (250 μL) were incubated with various concentrations of glucose (0–200 μM, 250 μL) at ambient temperature for 30 min. The fluorescence spectra of the resulting solutions were collected by operating the fluorescence spectrophotometer at an excitation wavelength of 380 nm.
Instrumentation
The absorption and fluorescence spectra were collected using a double-beam UV-vis spectrophotometer (Cintra 10e; GBC, Victoria, Australia) and a Hitachi F-7000 fluorometer (Hitachi, Tokyo, Japan), respectively. High-resolution transmission electron microscopy (HRTEM, FEI Tecnai G2 F20 S-Twin working at 200 kV) was used to take TEM images. The size and size distribution of particles were measured using dynamic light scattering (DLS) (N5 submicrometer particle size analyzer, Beckman Coulter Inc., U.S.A.). The lab-made dark-field microscopy (DFM) system consisted of an Olympus IX71 inverted microscope (Tokyo, Japan), a 100 W halogen lamp, a condenser (IX-ULWCD, Olympus, Japan), an objective (40×; numerical apertures = 0.75), and a digital camera (DP70, Olympus, Tokyo, Japan). Time-resolved fluorescence measurements of protein-stabilized AuNCs were performed using a time-correlated single photon counting system (Time-Harp 200, PicoQuant GmbH, Berlin, Germany) equipped with a pulsed diode laser at 390 nm (tens of ps pulse-width). The elemental compositions of protein-stabilized AuNCs were measured by JAMP-9500F Auger Electron Spectroscopy (JEOL, Japan). The quantification of gold was performed by inductively coupled plasma mass spectrometry (ICP-MS) (Perkin-Elmer-SCIEX, Thornhill, ON, Canada). A mass spectrometer MicrOTOF-QII from Bruker Daltonics (Billerica, MA, USA) was applied to obtain positive-ion electrospray ionization mass spectra. Raman spectra were measured using a confocal Raman microscope (CRM200, WITec, Ulm, Germany) equipped with a pulsed 532 nm laser. Native PAGE (Mini-PROTEAN Tetra Cell, BioRad, Richmond, CA) containing 7.5% separating gel and 4% (m/v) stacking gel was conducted at 120 V for 90 min. Gels were stained with a solution containing 0.04% w/v Coomassie brilliant blue R-250, 41% v/v methanol, and 9% v/v acetic acid for 1 h. After that, gels were de-stained with a solution containing 20% v/v methanol and 10% v/v acetic acid.3. Results and discussion
Reaction of Fenton reagent with GSH-AuNPs
GSH-AuNPs were produced by reducing HAuCl4 with NaBH4 in the presence of GSH. The as-prepared solution exhibited a surface plasmon resonance (SPR) at 525 nm, which is characteristic of AuNPs (Fig. 1A). The Fenton reaction of the GSH-AuNPs proceeded in an aqueous solution containing 100 mM H2O2 and 5 mM FeII. H2O2 can be dissociated into hydroxyl radicals via one-electron reduction by FeII.26 The reaction of the as-generated hydroxyl radicals and GSH-AuNPs was monitored over 60 min using absorption spectroscopy, TEM, and DLS. Fig. 1A reveals the gradual change in the intensity of the SPR with time, which accompanies a color change from purple to yellow. As a control experiment, the extinction spectra and solution color of GSH-AuNPs remained almost constant over time with the sole addition of FeII or H2O2 (Fig. S1 and S2, ESI†). The TEM image revealed that the average diameters of the GSH-AuNPs were 5(±2) nm (Fig. S3A, ESI†). After treatment of GSH-AuNPs with Fenton reagent for 1 h, the size of the resulting products was smaller than 5 nm (Fig. S3B, ESI†). The analysis of GSH-AuNPs and the resulting products by DLS showed that the hydrodynamic size of the resulting products was obviously smaller than that of GSH-AuNPs (Fig. S4, ESI†); this provides evidence that the hydroxyl radicals indeed etch the AuNPs at ambient temperature. Note that oligomeric Au–thiolate complexes did not form when GSH-AuNPs reacted with Fenton reagent for 1 h. | ||
| Fig. 1 Time-evolution of visible spectra of GSH-AuNPs in the presence of in the presence of 5 mM FeII and 100 mM H2O2. GSH-AuNPs were incubated with Fenton's solution at ambient temperature. | ||
When the reaction continued to 3 h, the final products displayed a broad absorption and no SPR band (Fig. 2A). This result is similar to that observed in oligomeric Au–thiolate complexes, such as sodium aurothiomalate (Myocrisin) and aurothioglucose (Solganal).27 Comparison of the Raman spectra of the GSH-AuNPs and final products revealed the appearance of characteristic peaks for the Au–S bonds at 225 and 286 cm−1; these peaks are associated with gold–sulfur vibrations, as reported by Varnholt et al.28 (Fig. 2B). The valence states of the GSH-AuNPs and final products were determined from their X-ray photoelectron spectroscopy (XPS) spectra. The XPS spectrum of the GSH-AuNPs exhibited two peaks at 87.6 and 83.9 eV, corresponding to Au 4f5/2 and Au 4f7/2, respectively. Deconvolution of the Au 4f7/2 spectrum resulted in two distinct components at 83.9 and 84.8 eV, which correspond to Au0 and AuI, respectively. The best fit of the data indicates that the GSH-AuNPs contained 4.6% AuI and 95.4% Au0 (curve a in Fig. 2C). In contrast, the binding energies of the Au 4f5/2 and Au 4f7/2 electrons in the final products were 88.5 and 84.8 eV, respectively, signifying that only AuI was present in the final products (curve b in Fig. 2C). Positive-ion ESI mass spectroscopy with the soft ionization technique was used to identify the composition of the final products. The peaks observed at m/z 770.4, 778.1 and 780.3 Da are consistent with [3Au197 + 5S32 + H2O16]+, [3Au197 + S32 + 4S34 + H2O16]+, and [3Au197 + 5S34 + H2O16]+ fragments, respectively (Fig. 2D). However, there is no peak in the ESI-MS spectrum that corresponds to the GSH-protected gold clusters (AunGSHm). Because GSH was prone to decomposition under hydroxyl-radical treatment, we suggest that most of the GSH decomposed and lost its thiol groups in the presence of the hydroxyl radicals. As a result, the final products contained sulfur instead of GSH. Similarly, previous studies demonstrated that GSH decomposed under thermal treatment, and the produced sulfur was used as a source for the synthesis of CdS, ZnS, and Ag2S quantum dots.29–33 According to previously reported results and our findings, we strongly believe that hydroxyl radical-induced etching of GSH-AuNCs produces oligomeric AuI–thiolate complexes rather than oligomeric Au0–thiolate complexes and GSH-protected Au clusters.
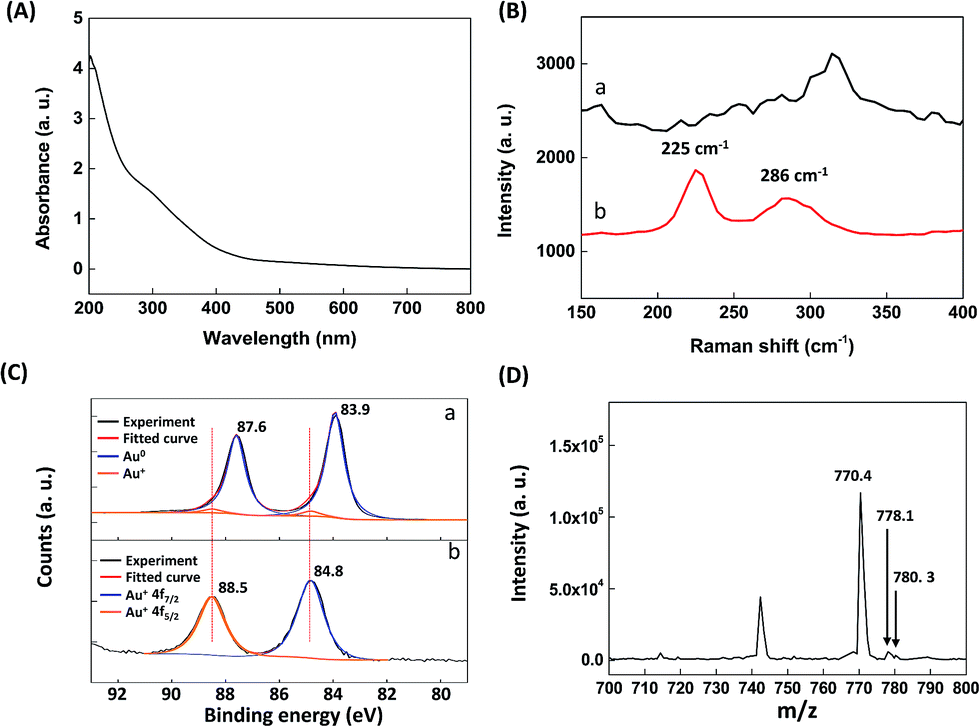 | ||
| Fig. 2 (A) Absorption spectrum and (D) ESI-MS spectrum of oligomeric AuI–thiolate complexes. (B) Raman spectra and (C) XPS spectra of (a) GSH-AuNPs and (b) oligomeric AuI–thiolate complexes. (C) (a) The original spectrum is in black, the fitted spectrum is in red, and the Au0 4f spectrum is in blue, and the AuI 4f spectrum is in orange. (b) The original spectrum is in black, the fitted spectrum is in red, and the AuI 4f7/2 spectrum is in blue, and the AuI 4f5/2 spectrum is in orange. | ||
Reaction of Fenton reagent with gold-based nanomaterials
DFM was used to monitor the hydroxyl radical-induced etching of a single gold nanoparticle with a diameter of 100 nm. Previous studies have demonstrated that 100, 50, and 10 nm single gold nanoparticles scatter red, green, and no light, respectively.34 Incubation of 100 nm gold nanoparticles with hydroxyl radicals at ambient temperature changed the intense red spots in the DFM images of the single gold nanoparticle to dim green spots, which vanished over time (Fig. S5, ESI†). Taken together, the scattering images of particles at the different etching stages provide further evidence of hydroxyl radical-induced etching of GSH-AuNPs.The successful etching of GSH-AuNSs by hydroxyl radicals suggests that the same strategy may be implemented to etch other gold-based nanomaterials. Short gold nanorods and octahedral gold nanocrystals, for example, were used to test this hypothesis. Compared to the treatment of GSH-AuNSs with hydroxyl radicals, a similar transition in the absorption spectra was observed in the treatment of short gold nanorods and octahedral nanocrystals with hydroxyl radicals (Fig. 3). TEM images show that the size of short gold nanorods and octahedral nanocrystals became extremely small (∼2 nm) after the addition of hydroxyl radicals, reflecting that hydroxyl radicals indeed etch gold nanorods and octahedral nanocrystals (Fig. S6 and S7, ESI†).
 | ||
| Fig. 3 Time evolution of visible spectra and photo images of (A) gold nanorods and (B) octahedral gold nanocrystals in the presence of 5 mM FeII and 100 mM H2O2. Gold-based nanomaterials were incubated with Fenton's solution at ambient temperature. The arrow indicates the direction of time evolution ((A) 0, 1, 3, 5, 10, 20, 30, 40, 50, and 60 min; (B) 0, 1, 3, 5, 10, 20, 30, 40, 50, and 60 min). | ||
Encouraged by these results, we envisaged that hydroxyl radicals could be used in place of cyanide for the removal of a layer of gold from scrap electronics. Importantly, the use of cyanide as a stripping solution has negative environmental implications. The gold-plated scrap was immersed into a solution of 100 mM H2O2 and 5 mM FeII at ambient temperature. ICP-MS was utilized to quantitatively determine the concentration of liberated gold ions in the solution after varied incubation times. The concentration of gold liberated from the scrap gradually increased over time (Fig. S8, ESI†), signifying that the hydroxyl radicals effectively remove gold from scrap electronics. We suggest that the efficiency of the hydroxyl radical-induced removal of gold from scrap electronics can be further improved by increasing the concentration of the Fenton's solution.
Protein-induced the aggregation of AuI–thiolate complexes
Many proteins, such as BSA, lysozyme, transferrin-family proteins, horseradish peroxidase, ribonuclease A, insulin, and trypsin, have been successfully used as templates for the preparation of red-emitting Au25 clusters under alkaline conditions.10,35 However, proteins are unable to retain their activity in protein-stabilized AuNCs because the synthetic process is conducted under a high concentration of NaOH. Although pepsin-stabilized AuNCs with blue (Au5 and Au8), green (Au13), and red (Au25) emissions were successfully prepared at pH values of 9.0, 1.0, and 12.0, respectively,36 there have been no reports on the synthesis of protein-stabilized AuNCs with different emissions under neutral conditions. Accordingly, a simple, one-pot, green synthetic route for preparing protein-stabilized AuNCs with different emissions is proposed in this study. The as-generated oligomeric AuI–thiolate complexes were directly mixed with different kinds of proteins at ambient temperature for ∼8 h. Because oligomeric AuI–thiolate complexes are relatively insoluble in aqueous solutions, they could move into hydrophobic pockets in the presence of proteins. This behavior could promote aggregation of the oligomeric AuI–thiolate complexes through intra- and intercomplex aurophilic interactions between AuI ions, leading to the formation of either Au0–thiolate clusters or AuI@Au0–thiolate core–shell clusters.As a proof-of-concept, three model proteins, including trypsin, lysozyme, and glucose oxidase, were selected to induce aggregation of the oligomeric AuI–thiolate complexes. The absorption spectra of the three as-synthesized samples are featureless (Fig. S9, ESI†), indicating that the proteins did not cause the formation of gold nanoparticles. After incubating the oligomeric AuI–thiolate complexes with trypsin, lysozyme, and glucose oxidase, the resulting products exhibited emission maxima at 415, 460, and 535 nm with excitation wavelengths of 350, 350, and 380 nm, respectively (Fig. 4A). The trypsin-, lysozyme-, and glucose oxidase-stabilized products emitted intense violet, blue, and green light, respectively. Fig. S10 (ESI)† demonstrates that the strong emissions arose from the final products rather than from the native fluorescence of the protein. The XPS spectra of trypsin-, lysozyme-, and glucose oxidase-stabilized products showed Au0 4f7/2 binding energies at 84.3, 84.0, and 83.9 eV, respectively (Fig. 4B). This shift is attributed to the fact that the binding energy of AuNCs increases with decreasing cluster size.36,37 Deconvolution of the Au 4f7/2 spectrum revealed that the trypsin- and lysozyme-stabilized products had very little AuI on the surface of the Au core, while the glucose oxidase-stabilized products contained approximately 70% Au0 and 30% AuI. A previous study reported that HgII efficiently quenched the fluorescence of protein-stabilized Au25 clusters through metallophilic bonding between HgII and AuI in Au25 clusters.38,39 Therefore, the presence of AuI in glucose oxidase-stabilized AuNCs could be confirmed by the addition of HgII. Fig. S11 (ESI)† shows that the fluorescence of glucose oxidase-stabilized AuNCs was quenched upon the addition of HgII, indicating the presence of AuI in the glucose oxidase-stabilized AuNCs. Based on the simple relationship (EFermi/N1/3) of the spherical Jellium model40 and the aforementioned results, the emissions at 415, 460, and 535 nm of the trypsin-, lysozyme-, and glucose oxidase-stabilized products, respectively, were identified to be derived from Au5–, Au8–, and Au13–thiolate clusters. The quantum yields of the Au5–, Au8–, and Au13–thiolate clusters calibrated with quinine sulfate were 2.9, 4.7, and 2.1%, respectively.
 | ||
| Fig. 4 (A) Fluorescence spectra of (a) trypsin-, (b) lysozyme-, and (c) glucose oxidase-stabilized AuNCs under the excitation wavelength at (a) 350, (b) 350, and (c) 380 nm. Digital photos of protein-stabilized AuNCs under visible and UV light. (B) XPS spectra of (a) trypsin-, (b) lysozyme-, and (c) glucose oxidase-stabilized AuNCs. (a–c) The original spectrum is in black, the Au0 4f7/2 spectrum is in red, and the Au0 4f5/2 spectrum is in blue, the AuI 4f7/2 spectrum is in orange, and the AuI 4f5/2 spectrum is in green. (C, E, and G) PAGE gels of protein (lane I) and protein-stabilized AuNCs (lane II): (C) trypsin, (E) lysozyme, and (G) glucose oxidase. (D, F, H) Fluorescence spectra of band 1, 2, and 3. | ||
Native polyacrylamide gel electrophoresis (PAGE) was used to confirm the formation of AuNCs from protein-induced aggregation of the oligomeric AuI–thiolate complexes. In contrast to the native-PAGE electropherograms of the three proteins (Fig. 4C, E and F, lane I), the native-PAGE electropherograms of the trypsin-, lysozyme-, and glucose oxidase-stabilized AuNCs all exhibited multiple bands as a result of protein oligomerization during the synthetic process (Fig. 4C, E and F, lane II). The multiple bands in the PAGE electropherograms were consistent with the analysis of human serum albumin-stabilized AuNCs by native PAGE.41 Because the molecular weight of trypsin resembles that of trypsin-stabilized Au8 clusters, we proposed that trypsin and trypsin-stabilized Au8 clusters could have similar charge-to-mass ratios. Accordingly, band 1 in lane II could be identical to that of trypsin-stabilized Au8 clusters. To prove this, band 1 was cut from the gel (Fig. 4C) and analyzed by fluorescence spectroscopy. The fluorescence spectrum of band 1 was almost the same as that of a solution of trypsin-stabilized Au5 clusters (Fig. 4D). Similarly, the fluorescence spectra of bands 2 and 3 resembled those of lysozyme- and glucose oxidase-stabilized AuNCs, respectively (Fig. 4F and H). These results support that the oligomeric AuI–thiolate complexes were assembled and converted to AuNCs inside the proteins. The stability of the protein-stabilized AuNCs was examined by monitoring their fluorescence under various conditions. The three protein-stabilized AuNCs exhibited excellent stability after storage in 10 mM phosphate (pH 6.0) at ambient temperature (Fig. S12, ESI†) and in the presence of a high salt concentration (Fig. S13, ESI†). This strategy is also suitable for the preparation of fluorescent AuNCs using other proteins, including bovine serum albumin (Fig. S14, ESI†) and choline oxidase (Fig. S15, ESI†).
Sensing of GSH and glucose
Shichibu et al. reported that GSH can etch AuNC clusters smaller than Au25.42 Thus, it is assumed that GSH could switch off the fluorescence of trypsin-, lysozyme-, and glucose oxidase-stabilized AuNCs through GSH-induced core-etching of AuNCs. Incubation of trypsin-stabilized AuNCs with 0–5 mM GSH at ambient temperature for 15 min resulted in a progressive decrease in the fluorescence of trypsin-stabilized AuNCs with increasing GSH concentration (Fig. 5A). The linear relationship (R2 = 0.9846) of the fluorescence intensity at 415 nm versus the GSH concentration ranged from 0.2 to 1 mM (Fig. 5B). The limit of detection (LOD) at a signal-to-noise ratio of three for GSH was estimated to be 60 μM, which is lower than the normal level (0.5–10 mM) of GSH in erythrocytes. As expected, GSH was capable of etching lysozyme- and glucose oxidase-stabilized AuNCs, thereby quenching their fluorescence because they are smaller than Au25 clusters (Fig. S16 and S17, ESI†). | ||
| Fig. 5 (A) Fluorescence intensity at 415 nm of trypsin-stabilized AuNCs in the presence of different concentrations of GSH. Trypsin-stabilized AuNCs were incubated with GSH in 10 mM phosphate (pH 6.0) at ambient temperature for 15 min. (B) A plot of the fluorescence intensity at 415 nm versus the GSH concentration. The error bars represent standard deviations based on three independent measurements. (C) Fluorescence intensity at 535 nm of glucose oxidase-stabilized AuNCs in the presence of different concentrations of glucose. Glucose oxidase-stabilized AuNCs were incubated with glucose in 10 mM phosphate (pH 6.0) at ambient temperature for 30 min. (D) A plot of the fluorescence intensity at 535 nm versus the glucose concentration. The error bars represent standard deviations based on three independent measurements. | ||
Jin et al. showed that H2O2 efficiently switched off the fluorescence of BSA-stabilized Au25 clusters through H2O2-induced oxidation of AuNCs.43 In addition, glucose oxidase catalyzes O2-mediated oxidation of glucose to form H2O2 and gluconic acid. Because the synthesis of glucose oxidase-stabilized Au13 clusters proceeded under neutral conditions, we hypothesized that glucose oxidase retains its activity in AuNCs. Thus, the fluorescence of glucose oxidase-stabilized Au13 clusters was monitored in the presence of variable glucose concentrations at pH 6.0 and recorded after a fixed time interval of 30 min. As the concentration of glucose increased, the fluorescence of the glucose oxidase-stabilized Au13 clusters was gradually quenched (Fig. 5C); this result provides clear evidence that glucose oxidase in AuNCs can catalyze the oxidation of glucose in the presence of O2, resulting in the production of H2O2. By plotting the fluorescence intensity at 535 nm against the glucose concentration, a linear range (R2 = 0.9862) for the quantification of glucose was observed from 2 to 40 μM (Fig. 5D). The LOD of glucose was determined to be 0.6 μM. Because the concentrations of tear glucose in normal and diabetic people are 0.2 ± 0.1 and 0.92 ± 0.52 mM, respectively,44 we suggest that the sensitivity of glucose oxidase-stabilized Au13 clusters is suitable for non-invasive monitoring of glucose in tear fluid.45
4. Conclusions
The present study demonstrated that hydroxyl radicals efficiently etch GSH-AuNPs to generate oligomeric AuI–thiolate complexes. The Fenton reaction could be applied for the removal of gold from scrap electronics. In addition, trypsin-, lysozyme-, and glucose oxidase-induced aggregation of non-fluorescent oligomeric AuI–thiolate complexes produced violet-emitting Au5, blue-emitting Au8, and green-emitting Au13 clusters, respectively. The applications of trypsin- and glucose oxidase-stabilized AuNCs for sensing GSH and glucose, respectively, were explored, and should pave the way for applications of protein-stabilized AuNCs in the field of biosensors. Interestingly, it was found that the oligomeric AuI–thiolate complexes can efficiently catalyze NaBH4-mediated reduction of 4-nitrophenol to 4-aminophenol (Fig. S18, ESI†). Further investigation of these results will follow.Acknowledgements
We would like to thank the Ministry of Science and Technology (NSC 100-2628-M-110-001-MY4) and for the financial support of this study.Notes and references
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed
.
- J. Zheng, C. Zhang and R. M. Dickson, Phys. Rev. Lett., 2004, 93, 077402 CrossRef
- S. H. Yau, O. Varnavski and T. Goodson, Acc. Chem. Res., 2013, 46, 1506–1516 CrossRef CAS PubMed
- Y. Lu and W. Chen, Chem. Soc. Rev., 2012, 41, 3594–3623 RSC
- L. Shang, F. Stockmar, N. Azadfar and G. U. Nienhaus, Angew. Chem., Int. Ed., 2013, 52, 11154–11157 CrossRef CAS PubMed
- L. Shang, L. Yang, F. Stockmar, R. Popescu, V. Trouillet, M. Bruns, D. Gerthsen and G. U. Nienhaus, Nanoscale, 2012, 4, 4155–4160 RSC
- X. Yuan, B. Zhang, Z. Luo, Q. Yao, D. T. Leong, N. Yan and J. Xie, Angew. Chem., Int. Ed., 2014, 53, 4623–4627 CrossRef CAS PubMed
- Z. Wang, W. Cai and J. Sui, ChemPhysChem, 2009, 10, 2012–2015 CrossRef CAS PubMed
- J. Zheng, J. T. Petty and R. M. Dickson, J. Am. Chem. Soc., 2003, 125, 7780–7781 CrossRef CAS PubMed
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS PubMed
- T. Wang, X. Hu and S. Dong, Chem. Commun., 2008, 4625–4627 RSC
- S. Riaz, W. Ma, C. Jing, M. H. Nawaz, D. W. Li and Y. T. Long, Chem. Commun., 2013, 49, 1738–1740 RSC
- C. Y. Ke, T. H. Chen, L. C. Lu and W. L. Tseng, RSC Adv., 2014, 4, 26050–26056 RSC
- C. C. Huang, Z. Yang, K. H. Lee and H. T. Chang, Angew. Chem., Int. Ed., 2007, 46, 6824–6828 CrossRef CAS PubMed
- C. A. Lin, T. Y. Yang, C. H. Lee, S. H. Huang, R. A. Sperling, M. Zanella, J. K. Li, J. L. Shen, H. H. Wang, H. I. Yeh, W. J. Parak and W. H. Chang, ACS Nano, 2009, 3, 395–401 CrossRef CAS PubMed
- C. C. Huang, C. T. Chen, Y. C. Shiang, Z. H. Lin and H. T. Chang, Anal. Chem., 2009, 81, 875–882 CrossRef CAS PubMed
- M. H. Muhammed, S. Ramesh, S. P. S. Sinha and T. Pradeep, Nano Res., 2008, 1, 333–340 CrossRef CAS
- E. Neyens and J. Baeyens, J. Hazard. Mater., 2003, 98, 33–50 CrossRef CAS
- J. J. Pignatello, E. Oliveros and A. MacKay, Crit. Rev. Environ. Sci. Technol., 2006, 36, 1–84 CrossRef CAS PubMed
- H. N. Quan, A. L. Teel and R. J. Watts, J. Hazard. Mater., 2003, 102, 277–289 CrossRef CAS
- J. Yang, L. Hong, Y. H. Liu, J. W. Guo and L. F. Lin, Environ. Technol., 2014, 35, 2878–2884 CrossRef CAS PubMed
- P. Krystynik, P. Kluson, S. Hejda, P. Masin and D. N. Tito, Water Environ. Res., 2014, 86, 2212–2220 CrossRef CAS
- P. E. Karthik, C. Jeyabharathi and K. L. Phani, Chem. Commun., 2014, 50, 2787–2790 RSC
- A. M. Nowicka, U. Hasse, M. Hermes and F. Scholz, Angew. Chem., Int. Ed., 2010, 49, 1061–1063 CrossRef CAS PubMed
- X. Zhou, Y. Zhang, C. Wang, X. Wu, Y. Yang, B. Zheng, H. Wu, S. Guo and J. Zhang, ACS Nano, 2012, 6, 6592–6599 CrossRef CAS PubMed
- S. H. Bossmann, E. Oliveros, M. Kantor, S. Niebler, A. Bonfill, N. Shahin, M. Worner and A. M. Braun, Water Sci. Technol., 2004, 49, 75–80 CAS
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS PubMed
- B. Varnholt, P. Oulevey, S. Luber, C. Kumara, A. Dass and T. Bürgi, J. Phys. Chem. C, 2014, 118, 9604–9611 CAS
- P. Huang, Q. Jiang, P. Yu, L. Yang and L. Mao, ACS Appl. Mater. Interfaces, 2013, 5, 5239–5246 CAS
- Y. Zheng, S. Gao and J. Y. Ying, Adv. Mater., 2007, 19, 376–380 CrossRef CAS PubMed
- Q. Wu, H. Cao, S. Zhang, X. Zhang and D. Rabinovich, Inorg. Chem., 2006, 45, 7316–7322 CrossRef CAS PubMed
- Y. F. Liu and J. S. Yu, J. Colloid Interface Sci., 2010, 351, 1–9 CrossRef CAS PubMed
- L. Tan, A. Wan and H. Li, ACS Appl. Mater. Interfaces, 2013, 5, 11163–11171 CAS
- C. Inhee and C. Yeonho, IEEE J. Sel. Top. Quantum Electron., 2012, 18, 1110–1121 CrossRef
- L. Y. Chen, C. W. Wang, Z. Yuan and H. T. Chang, Anal. Chem., 2015, 87, 216–229 CrossRef CAS PubMed
- H. Kawasaki, K. Hamaguchi, I. Osaka and R. Arakawa, Adv. Funct. Mater., 2011, 21, 3508–3515 CrossRef CAS PubMed
- X. L. Guével, B. Hötzer, G. Jung, K. Hollemeyer, V. Trouillet and M. Schneider, J. Phys. Chem. C, 2011, 115, 10955–10963 Search PubMed
- Y. H. Lin and W. L. Tseng, Anal. Chem., 2010, 82, 9194–9200 CrossRef CAS PubMed
- J. P. Xie, Y. G. Zheng and J. Y. Ying, Chem. Commun., 2010, 46, 961–963 RSC
- J. Zheng, P. R. Nicovich and R. M. Dickson, Annu. Rev. Phys. Chem., 2007, 58, 409–431 CrossRef CAS PubMed
- Z. Zheng, Y. Zhou, X. Li, S. Liu and Z. Tang, Biosens. Bioelectron., 2011, 26, 3081–3085 CrossRef CAS PubMed
- Y. Shichibu, Y. Negishi, H. Tsunoyama, M. Kanehara, T. Teranishi and T. Tsukuda, Small, 2007, 3, 835–839 CrossRef CAS PubMed
- L. Jin, L. Shang, S. Guo, Y. Fang, D. Wen, L. Wang, J. Yin and S. Dong, Biosens. Bioelectron., 2011, 26, 1965–1969 CrossRef CAS PubMed
- D. K. Sen and G. S. Sarin, Br. J. Ophthalmol., 1980, 64, 693–695 CrossRef CAS
- V. L. Alexeev, S. Das, D. N. Finegold and S. A. Asher, Clin. Chem., 2004, 50, 2353–2360 CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures for prepared compounds and Fig. S1–S15. See DOI: 10.1039/c5ra05159b |
| This journal is © The Royal Society of Chemistry 2015 |