
Quaternary structures of GroEL and naïve-Hsp60 chaperonins in solution: a combined SAXS-MD study†
Abstract
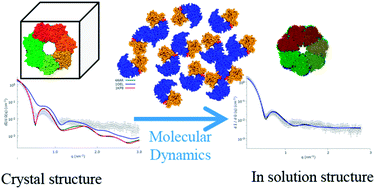
The quaternary structures of bacterial GroEL and human naïve-Hsp60 chaperonins in physiological conditions have been investigated by an innovative approach based on a combination of synchrotron Small Angle X-ray Scattering (SAXS) in-solution experiments and molecular dynamics (MD) simulations. Low-resolution SAXS experiments over large and highly symmetric oligomers are analyzed on the basis of the high-resolution structure of the asymmetric protein monomers, provided by MD. The results reveal remarkable differences between the solution and the crystallographic structure of GroEL and between the solution structures of GroEL and of its human homologue Hsp60.
 Please wait while we load your content...
Please wait while we load your content...

