
Synthesis and characterization of ZnO nanorods with a narrow size distribution†
Abstract
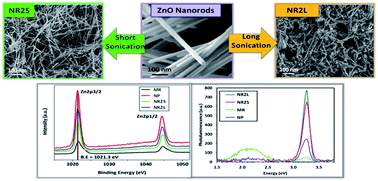
The development of novel materials for energy harvesting applications or strain sensing has generated great interest towards zinc oxide (ZnO) nanostructures, and in particular towards the synthesis of ZnO nanowires or nanorods with well controlled morphology and properties. The high-yield mass production of such nanostructures by catalyst-free methods is a crucial aspect to enable a cost-effective large-scale development of new ZnO-based piezoelectric devices and materials. In the present work, we propose a method for the mass-production of high-purity ZnO-nanorods with a uniform size distribution, based on the combination of thermal decomposition of zinc acetate dihydrate and probe sonication in acetone. The quality of the produced ZnO nanorods is assessed through multi-technique characterization using field-emission scanning electron microscopy (FE-SEM), X-ray diffraction (XRD), transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), and photo-luminescence spectroscopy (PL). The adopted synthesis method is simple, cost effective and feasible for large-scale production. Various process parameters such as precursor amount and growth time have been found to play an important role in controlling the formation of the as grown nanostructures with high uniformity in size and morphology. Size distribution curves were employed to depict the effect of various process parameters for tailoring the morphology, homogeneity and aspect ratio of the nanorods. Our results reveal that the high crystallographic quality of ZnO nanorods grown by a long-time thermal decomposition method is not affected by probe sonication, which is proposed as a post-synthesis step necessary to produce ZnO nanorod powder with a uniform distribution of diameters and lengths.
 Please wait while we load your content...
Please wait while we load your content...

