
One-pot synthesis of magnetite-loaded dual-mesoporous silica spheres for T2-weighted magnetic resonance imaging and drug delivery†
Xiaofeng Luoa,
Dechao Niu*a,
Yao Wanga,
Yungang Zhaia,
Jianzhuang Chena,
Jinlou Gua,
Jianlin Shiab and
Yongsheng Li*a
aLab of Low-Dimensional Materials Chemistry, School of Materials Science and Engineering, East China University of Science and Technology, Shanghai, 200237, China. E-mail: ysli@ecust.edu.cn; dcniu@ecust.edu.cn; Tel: +86-21-64250740
bState Key Laboratory of High Performance Ceramics and Superfine Microstructures, Shanghai Institute of Ceramics, Chinese Academy of Sciences, 1295 Ding-xi Road, Shanghai, 200050, China
First published on 24th April 2015
Abstract
The combination of mesoporous silica nanoparticles and superparamagnetic nanocrystals to fabricate multifunctional platforms presents great potentials for simultaneous imaging and drug delivery. In this work, we have successfully developed a simple one-step approach to synthesize magnetite-loaded dual-mesoporous silica spheres consisting of large pores in the core and small pores in the shell (Fe3O4@DMSSs) by embedding oil-soluble Fe3O4 into the large pores of DMSSs, which were prepared by employing polystyrene-b-poly(acrylic acid) (PS-b-PAA) and cetyl trimethyl ammonium bromide (CTAB) as dual-templates. The loading amounts of magnetite can be easily adjusted by varying the initial concentrations of Fe3O4 nanoparticles in the oil phase. The in vitro test indicates that Fe3O4@DMSSs possesses excellent T2-weighted magnetic resonance (MR) imaging performance with a maximum T2 relaxivity (r2) of 421.5 mMFe−1 S−1. Furthermore, a high doxorubicin (DOX) loading capacity (65 wt%) was achieved and the obtained DOX-loaded Fe3O4@DMSSs (DOX/Fe3O4@DMSSs) exhibits pH-sensitive behaviour with accelerated release of DOX in acidic environment. Confocal laser scanning microscopy observation shows that DOX/Fe3O4@DMSSs was able to locate in the cytoplasm of MCF-7 cells and release DOX into the nucleus to kill cancer cells. Therefore, it is anticipated that Fe3O4@DMSSs can be promising candidates as both T2-weighted MR contrast agents and drug delivery carriers in further biomedical applications.
Introduction
With the rapid development of nanotechnology, great attention and efforts have been devoted to the design and fabrication of various types of nanomaterials for biomedical applications, such as imaging, diagnosis and therapy.1–4 Among these nanomaterials, magnetic nanoparticles, especially for iron oxide nanoparticles, a unique class of functional nanocrystals that plays an important role in research and development of wide application fields such as magnetic fluids, bio-separation and magnetic resonance imaging (MRI), have attracted intensive attention owing to their excellent magnetism, good biocompatibility, tunable sizes and increased contrast enhancement.5–8 To date, several methods have been developed for synthesizing superparamagnetic iron oxide nanoparticles, such as co-precipitation,9 microemulsion,10 thermal decomposition11,12 and hydrothermal treatment.13 Among these, thermal decomposition has been widely employed to synthesize iron oxide nanoparticles as MRI contrast agents owing to their controllable size, monodispersity and high crystallinity.14 However, the hydrophobic feature of thus obtained magnetic nanoparticles greatly limits its applications in biomedicine. In order to improve its dispersion and stability in aqueous systems, various materials such as polyethylene glycol (PEG),15 dimercaptosuccinic acid (DMSA),16 amphiphilic polymers17,18 and silica19,20 have been employed for coating and stabilizing magnetic nanoparticles to extend their applications.Mesoporous silica nanoparticles (MSNs), as one of the most promising inorganic drug delivery carriers, have been widely investigated due to their unique physicochemical properties including high specific surface area and large pore volume, tunable pore sizes and morphologies, good biocompatibility and easily modified outer/inner surfaces.21–24 Recently, the combination of MSNs and iron oxide nanoparticles to prepare magnetic mesoporous silica nanoparticles (M-MSNs) has been widely exploited due to their potentials for simultaneous MRI and drug delivery.25–28 Hyeon et al. reported the synthesis of core–shell MSNs by using single Fe3O4 nanocrystals as cores (Fe3O4@mSiO2) and demonstrated their good performance in T2-weighted MRI and drug delivery.25 J. Zink et al. described the fabrication of multifunctional nanoparticles with several iron oxide nanocrystals encapsulated within MSNs and anticancer drugs stored inside the pores, and the materials exhibited great potential in simultaneous imaging and therapeutic applications.27 Recently, Shi et al. reported the fabrication of a multifunctional platform for bio-imaging and anticancer drug delivery by integrating a hollow iron oxide nanocapsule with a mesoporous silica shell.29 Unfortunately, these M-MSNs always have relatively low transverse relaxivity (r2 < 300 mMFe−1 S−1) as single or fewer superparamagnetic iron oxide nanoparticles have been loaded into the mesoporous structure. Moreover, the pore sizes of the reported M-MSNs are relatively small (2–5 nm), which limits the drug loading capability and treatment effect of M-MSNs. To address these issues, herein, we report a simple one-step approach to construct a novel nanocarrier platform for both high transverse relaxivity and efficient drug delivery based on magnetite-loaded dual-mesoporous silica spheres (Fe3O4@DMSSs), which consists of large pores in the core and small pores in the shell, by embedding multiple Fe3O4 nanoparticles into the large pores of DMSSs. To exploit its potential biomedical applications, the efficacy in T2-weighted magnetic resonance imaging (MRI), drug storage capacity and the cellular uptake and cancer therapy of Fe3O4@DMSSs were investigated.
Results and discussion
Fig. 1 shows the representative TEM images of Fe3O4@DMSSs prepared with different loading amounts of Fe3O4 nanoparticles (5, 15 and 30 mg) with average diameter of 6 nm (Fig. S1a†), denoted as Fe3O4@DMSSs-5, Fe3O4@DMSSs-15 and Fe3O4@DMSSs-30, respectively. As shown in Fig. 1a, it is clearly observed that well-defined and core–shell structured dual-mesoporous silica spheres with Fe3O4 nanoparticles (dark or black dots in Fig. 1b) encapsulated in the large pores in the core were obtained. On increasing the loading amount of Fe3O4 nanoparticles, more magnetic nanoparticles were found incorporated into the large pores (Fig. 1c and d). Further increasing it from 15 to 30 mg, almost all the large pores were filled with magnetic nanoparticles (Fig. 1e and f). In the XRD patterns, all the three Fe3O4@DMSSs present five distinct diffraction peaks in the range of 25–65° (Fig. S2†), which can be assigned to 220, 311, 400, 511 and 440 reflections of the Fe3O4 crystal phase with space group of Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (JCPDS card number: 19-0629) (Fig. S1b†), confirming the successful encapsulation of Fe3O4 nanoparticles in DMSSs. Moreover, the intensities of the diffraction peaks increase with the loading amounts of Fe3O4 nanoparticles, suggesting that more magnetite nanoparticles were embedded in the large pores.
m (JCPDS card number: 19-0629) (Fig. S1b†), confirming the successful encapsulation of Fe3O4 nanoparticles in DMSSs. Moreover, the intensities of the diffraction peaks increase with the loading amounts of Fe3O4 nanoparticles, suggesting that more magnetite nanoparticles were embedded in the large pores.
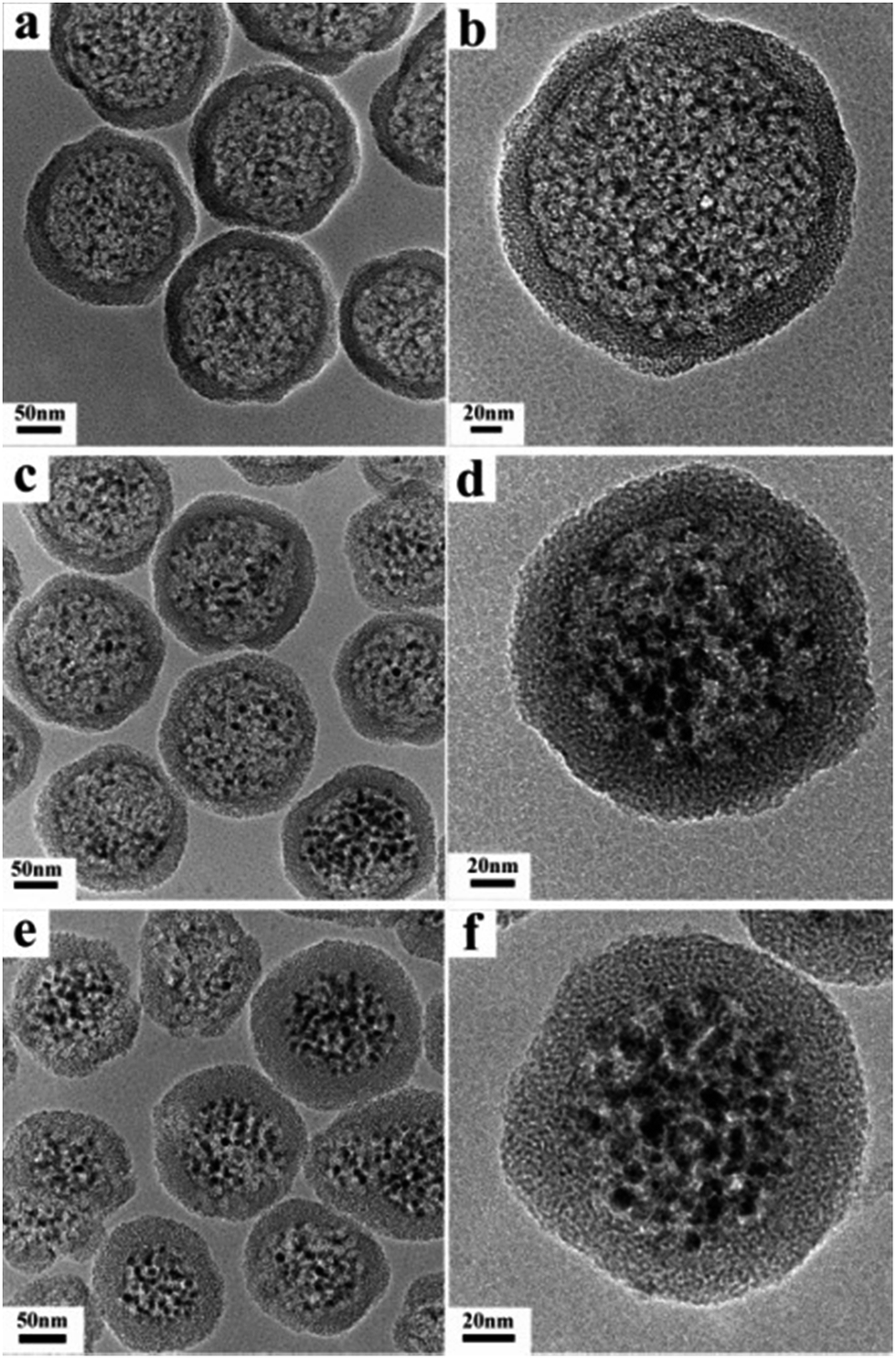 | ||
| Fig. 1 TEM images of Fe3O4@DMSSs-5 (a and b), Fe3O4@DMSSs-15 (c and d), and Fe3O4@DMSSs-30 (e and f). | ||
To investigate the effect of the introduction of Fe3O4 on the pore structure of DMSSs, N2 sorption analysis was conducted and the corresponding isotherms and pore size distribution curves are shown in Fig. 2. It is found that all the three samples exhibit type IV isotherms with two major capillary condensation steps at relative pressure of 0.2–0.3 and 0.85–0.95, respectively, implying the dual-mesoporous structure of the samples. Noticeably, the encapsulation of Fe3O4 nanoparticles did not change the core–shell dual-mesoporous structure of DMSSs, though the pore diameter distribution of larger pores becomes broader due to the introduction and accumulation of hydrophobic Fe3O4 nanoparticles within different large pores. Table 1 presents the synthetic and structural parameters of the samples. It is found that the specific surface area and pore volume are as high as 832 m2 g−1 and 1.12 cm3 g−1 for Fe3O4@DMSSs-5, 760 m2 g−1 and 1.03 cm3 g−1 for Fe3O4@DMSSs-15 and 842 m2 g−1 and 0.97 cm3 g−1 for Fe3O4@DMSSs-30, respectively. The loading amounts of Fe3O4 nanoparticles in the DMSSs determined by ICP analysis are about 4.8 wt% for Fe3O4@DMSSs-5, 13.3 wt% for Fe3O4@DMSSs-15 and 24.3 wt% for Fe3O4@DMSSs-30, respectively. In addition, the hydrodynamic diameter testing results by dynamic light scattering (DLS) (Fig. S3†) indicate that the particle size increases gradually with the adding amounts of magnetite particles. The mean particle size of the samples is ca. 234, 247 and 276 nm, respectively, with narrow particle distributions, demonstrating the good monodispersivity of Fe3O4@DMSSs in aqueous solution. These verify that monodisperse, dual mesoporous silica nanospheres loaded with Fe3O4 nanoparticles could be facilely fabricated with one-pot synthesis.
 | ||
| Fig. 2 N2 adsorption–desorption isotherms (A) and pore size distributions (B) of Fe3O4@DMSSs samples. (a) Fe3O4@DMSSs-5; (b) Fe3O4@DMSSs-15; (c) Fe3O4@DMSSs-30. | ||
| Sample | Fe3O4 amount (mg) | Loading amounts (wt%) | dsmall/nm | dlarge/nm | S/m2 g−1 | Vt/cm3 g−1 |
|---|---|---|---|---|---|---|
| Fe3O4@DMSSs-5 | 5 | 4.8 | 2.2 | 17/21.8 | 832 | 1.12 |
| Fe3O4@DMSSs-15 | 15 | 13.3 | 2.2 | 17/22 | 760 | 1.03 |
| Fe3O4@DMSSs-30 | 30 | 24.3 | 2.2 | 17 | 842 | 0.97 |
The magnetic properties of Fe3O4@DMSSs-5, Fe3O4@DMSSs-15 and Fe3O4@DMSSs-30 were measured by using vibrating sample magnetometer (VSM). As shown in Fig. S4,† the room-temperature magnetization curves show no hysteresis loop, demonstrating the superparamagnetic feature of all the three samples. In addition, the saturation magnetization values of these samples are calculated to be 1.5 emu per gram of Fe3O4@DMSSs-5, 3.6 emu per gram of Fe3O4@DMSSs-15 and 5.5 emu per gram of Fe3O4@DMSSs-30, respectively. To evaluate the T2-weighted MR imaging capability of Fe3O4@DMSSs, the transverse relaxation was measured by using a clinical 3.0 T MRI scanner and the transverse (r2 value) relaxivity was calculated through the curve fitting of 1/T2 relaxation time versus the Fe concentration (Fig. 3). The result shows that the r2 value increases with the increasing of Fe3O4 loading amount. It is calculated to be 370.2 mM mMFe−1 S−1 for Fe3O4@DMSSs-5, 385.8 mMFe−1 S−1 for Fe3O4@DMSSs-15 and 421.5 mMFe−1 S−1 for Fe3O4@DMSSs-30, respectively, demonstrating the excellent MR imaging capabilities of Fe3O4@DMSSs as a novel kind of T2-weighted MR contrast agent. Noticeably, the r2 values of Fe3O4@DMSSs are much higher than that of commercial iron oxide-based contrast agent (Feridex, r2 = 108 mMFe−1 S−1) and most of the iron oxide-based mesoporous nanoparticles.25–27,29 This is probably attributed to the dual-mesoporous structure, which is feasible for water molecules to contact magnetite nanoparticles, resulting in shortened transverse relaxation period and consequently intensified MR imaging performance.30 Besides, the synergetic effect17,18,31 between the multiple magnetite nanoparticles is also responsible for this.
 | ||
| Fig. 3 Plots of inverse transverse relaxation time (1/T2) versus Fe concentration of the samples. (a) Fe3O4@DMSSs-5, (b) Fe3O4@DMSSs-15, (c) Fe3O4@DMSSs-30. | ||
To explore the drug delivery performance of Fe3O4@DMSSs, doxorubicin (DOX), a chemotherapeutic drug was chosen as a model and loaded into the pores of Fe3O4@DMSSs-5 via the strong electrostatic interaction between the positively charged DOX and negatively charged pore channels. The DOX loading process can be monitored by UV-vis absorbance spectrometry. As shown in Fig. S5,† the intensity of the absorbance peak at 480 nm, which is the characteristic wavelength of DOX, decreases with the loading process, demonstrates the successful loading of DOX into Fe3O4@DMSSs-5. The DOX loading content was measured to be 65 wt%, which is much higher than that of the reported magnetic mesoporous silica nanoparticles.26,32 On the other hand, the significant decrease in both specific surface area and pore volume of DOX/Fe3O4@DMSSs-5 (44 m2 g−1 and 0.09 cm3 g−1) further indicates that most of the pores has been occupied by the adsorbed DOX drug molecules.
It is of great importance to investigate the drug release feature of Fe3O4@DMSSs as a practical drug delivery system for cancer chemotherapy. As a result, the in vitro DOX release properties of DOX/Fe3O4@DMSSs-5 were examined in PBS solutions at various pH values (Fig. 4). It is showed that the DOX release behaviour of DOX/Fe3O4@DMSSs-5 is pH-dependent. In details, in a pH = 7.4 solution, only about 8.5% of DOX was released within 18 h, which is vitally important to maintain the drugs less cytotoxic to the normal cells. By increasing the acidity of the solution to a pH value of 6.4, the release amount reached 18% within 24 h, and in a more acidic solution of pH 5.4, a sustained release followed by a fast release within the first 10 h was obtained, and about 40% of DOX was released from the nanocarriers in 72 h. This pH-responsive drug release behavior is attributed to the following two factors: (1) at lower pH values, the protonation of amine groups on DOX molecules (pKa = 8.2) become stronger, which could increase the hydrophilicity and solubility of DOX molecules;33 (2) the decreased pH would weaken the electrostatic interaction between negatively charged Fe3O4@DMSSs and positively charged DOX molecules, resulting in the faster release rate, which was similar with other reported literatures.34,35 Additionally, the pH dependent release character of the carriers may benefit the DOX release in relative acidic tumour microenvironment. Meanwhile, it is desirable that most DOX encapsulated in the pore channels would not leach out during in vivo circulation in the blood with a pH value of 7.4, and enable large amount of intracellular drug released once the nanoparticles are internalized inside the tumour cells by endocytosis as endosome/lysosome has a low pH value.36
 | ||
| Fig. 4 Release profiles of DOX/Fe3O4@DMSSs-5 at 37 °C under various pH values. | ||
In order to investigate the cellular uptake of Fe3O4@DMSSs, fluorescein isothiocyanate (FITC) was chosen as a marking dye to graft onto the surface of the nanoparticles (denoted as FITC-Fe3O4@DMSSs) via silane conjugation chemistry. The obtained FITC-Fe3O4@DMSSs were incubated with MCF-7 cells for 4 h at 37 °C in the culture medium, and analyzed by confocal laser scanning microscopy (CLSM). As shown in Fig. 5a, it is observed that FITC-Fe3O4@DMSSs were internalized by the MCF-7 cells and localized in the cytoplasm within 4 h of incubation. The internalization is clearly evident since the MCF-7 cell nucleus was stained with DAPI blue dye, suggesting the cellular uptake instead of adhesion to the surface of particles. In addition, the intracellular distribution of DOX/Fe3O4@DMSSs in MCF-7 cells was estimated. As shown in Fig. 5b, strong fluorescence was emitted from the cells treated with DOX/Fe3O4@DMSSs solution, particularly the nuclear regions after 4 h of incubation in MCF-7 cells, indicating that DOX/Fe3O4@DMSSs was efficiently internalized in MCF-7 cells by nonspecific endocytosis. Furthermore, Fe3O4@DMSSs could not be transported into the nuclei while free DOX released from DOX/Fe3O4@DMSSs was able to enter the nuclei by passive diffusion, which was demonstrated by the presence of strong red fluorescence emitted from nuclei of MCF-7 cells treated with DOX/Fe3O4@DMSSs. These may be attributed to the fast release of DOX in the low pH region in the cells (e.g. the pH values of endosomes and lysosomes are ca. 5.0–5.5),37 which is consistent with the in vitro release profiles of DOX/Fe3O4@DMSSs (Fig. 4). From the above observations, it is concluded that DOX/Fe3O4@DMSSs could penetrate into the living cells and thus the loaded DOX could be released from the nanocarriers.
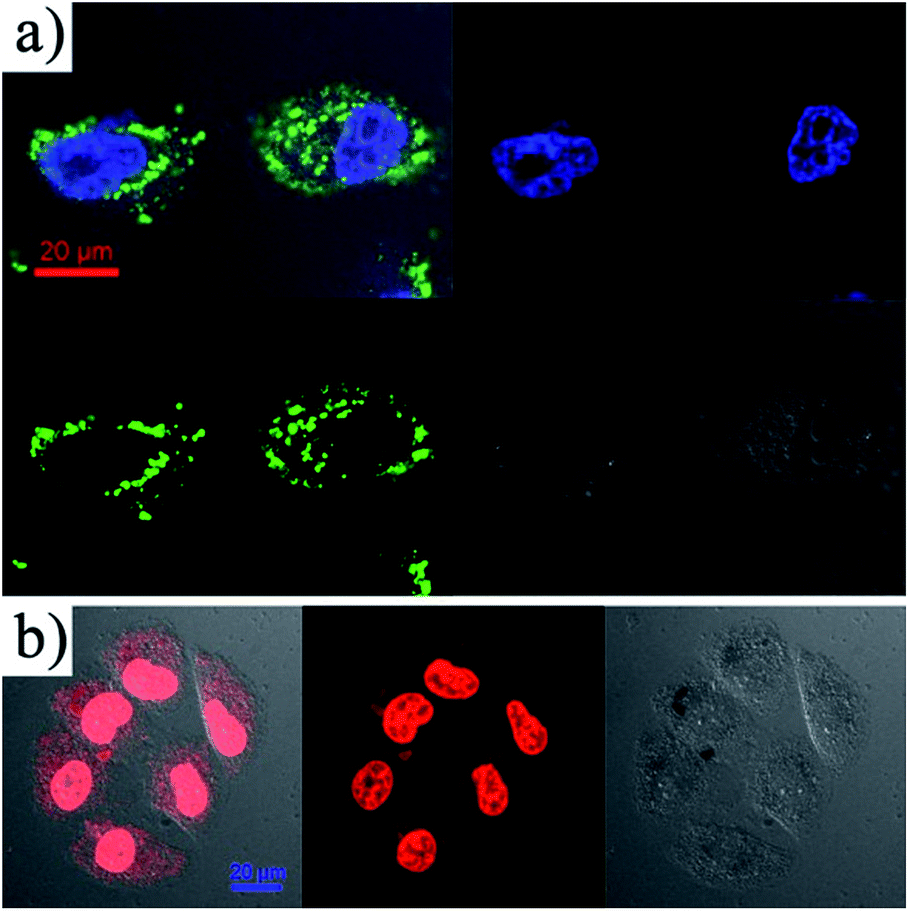 | ||
| Fig. 5 CLSM images of MCF-7 cells incubated with FITC-Fe3O4@DMSSs (a) and DOX/Fe3O4@DMSSs (b) for 4 h. | ||
To further verify whether the released DOX was still pharmacologically active, in vitro cytotoxicity tests against MCF-7 cells were investigated. Cell viabilities against DOX/Fe3O4@DMSSs and free DOX at different concentrations are shown in Fig. 6. The results reveal that the cytotoxic efficacy of the DOX/Fe3O4@DMSSs was comparable to free DOX after cultured with MCF-7 cells for 24 h. Moreover, more than half of tumor cells were effectively killed when incubated with DOX/Fe3O4@DMSSs for 24 h, indicating that DOX delivered by Fe3O4@DMSSs entered the MCF-7 cells and retained its pharmaceutical activity. In contrast, the DOX-free Fe3O4@DMSSs showed very low cytotoxicity against MCF-7 cells as well as representative normal cells (L02 normal human liver cells) (Fig. S6†). Consequently, Fe3O4@DMSSs has been proved to be one of promising nanocarrier candidates in drug loading and delivery in further cancer chemotherapy.
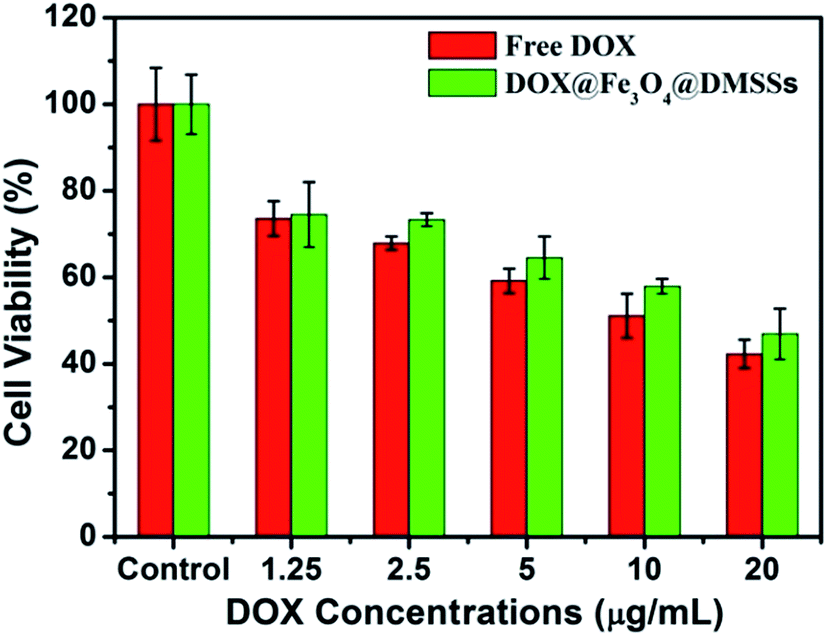 | ||
| Fig. 6 In vitro cytotoxicity of free DOX and DOX/Fe3O4@DMSSs against MCF-7 cells in 24 h of incubation. | ||
Conclusion
In summary, a one-step route has been successfully developed to synthesize magnetite-loaded dual-mesoporous silica spheres (Fe3O4@DMSSs) consisting of large pores in the core and small pores in the shell for T2-weighted magnetic resonance imaging and drug delivery. The obtained Fe3O4@DMSSs displays a well-fine core–shell morphology and good monodispersion. Moreover, Fe3O4@DMSSs presents excellent T2-weighted MR imaging effect with a high T2 relaxivity (r2 > 350 mMFe−1 S−1). In addition, Fe3O4@DMSSs shows high loading capacity (65 wt%) for doxorubicin due to its unique dual-mesoporous structure with high specific surface area and pore volume. More importantly, the cytotoxicity of DOX-loaded Fe3O4@DMSSs against MCF-7 cells is comparable to free DOX at relatively low drug concentrations owing to the intracellular release of drugs from DOX-loaded Fe3O4@DMSSs in cells. These unique properties endow them with great application potentials as anticancer drug carriers for the simultaneous imaging diagnosis and chemotherapy applications in future.Experimental section
Chemicals and materials
Cetyl trimethyl ammonium bromide (CTAB, ≥99%), ammonia solution (25–28%) and tetraethyl orthosilicate (TEOS, AR) were purchased from Shanghai Lingfeng Chemical Reagent Co. LTD. Tetrahydrofuran (THF, AR) and ethanol (AR) were purchased from Sinopharm Chemical Reagent Co. LTD (Shanghai, China). The pure water with a resistivity of 18.2 MΩ cm was used in all of experiments. All of reagents were used without further purification.Synthesis of PS100-b-PAA16 and magnetite nanoparticles
Amphiphilic block copolymer, polystyrene100-b-poly(acrylic acid)16 (PS100-b-PAA16), was synthesized via sequential atomic transfer radical polymerization (ATRP) as previously reported.38 Monodispersed 6 nm sized and hydrophobic Fe3O4 nanoparticles were prepared following the thermal decomposition method reported by Sun et al.11Synthesis of magnetite-loaded dual-mesoporous silica spheres (Fe3O4@DMSSs)
Fe3O4@DMSSs was prepared according to the previous method reported by our group.39 In a typical synthesis, 0.05 g of PS100-b-PAA16 and 5 mg of Fe3O4 were first dissolved in 10 mL of THF. Then the above oil solution was poured into a mixture solution containing 40 mL of H2O, 0.065 g of CTAB and 1.5 mL of ammonia. After that, 80 mL of ethanol was added into the mixture oil–water solution. After stirring for 2 h, 0.3 g of TEOS dissolved in 5 mL of ethanol was added into the above solution with continuous stirring in 0.5 h. After stirring for 18 h at room temperature, the as-synthesized sample was collected by centrifugation (1000 rpm, 10 min) and washed several times with water and ethanol. Finally, the product was obtained by dried in an oven and calcined at 550 °C for 6 h for surfactants removal.In vitro MR imaging
The in vitro MR imaging experiment was performed on a 3.0 T clinical MRI instrument (GE Signa HDx 3.0 T). For the T2-weighted fast-recovery fast spin-echo (FR-FSE) sequence, the following parameters were used: TR (repetition time) = 2000 ms, TE (echo time) = 107.1 ms, field of view (FOV) = 14 ms, slice thickness = 2.0 mm, echo length = 16, matrix = 256 × 192, number of acquisitions = 4. For T2 relaxivity measurement, firstly, the Fe concentration of the Fe3O4@DMSSs in water was determined by inductively coupled plasma atomic emission spectrometry (ICP-AES) after dissolving the samples in a mixture solution of HNO3/HClO4 at 150 °C. Then, solutions of samples containing different Fe concentrations were prepared in pure water. The T2 relaxation time was performed with the following parameters: TR = 4000 ms, TE = 13, 26, 39, 52 ms. Relaxivity values of r2 were calculated through the curve fitting of 1/T2 relaxation time (s−1) versus the Fe concentration (mM).DOX storage and release
Typically, 15 mg of DOX was completely dissolved in 10 mL of phosphate buffer solution (PBS). Then, the 20 mg of Fe3O4@DMSSs-5 was added into the DOX–PBS solution stirred for 24 h in dark at room temperature. The products were washed quickly several times with PBS to remove the physically adsorbed DOX residue on the surface and dried in a vacuum oven for 24 h. The concentration of the drug retained in the solution was determined by UV-vis spectrometer at 480 nm. The loading amount of the drug was calculated according to margin of the initial and the residual drug. In vitro drug release experiments were carried out in PBS at different pH values (5.4, 6.4 and 7.4). The DOX-loaded Fe3O4@DMSSs-5 (5 mg) were suspended in 3 mL PBS in the dialysis membrane bag (molecular weight cut-off 3500 Da) and the bag was immersed in 27 mL PBS and shaken at a speed of 100 rpm at 37 °C. At predetermined time intervals, 3.0 mL of the release buffer was removed from the tube, and then 3.0 mL of the fresh buffer was added to make the loss of solution. The collected buffer samples were examined by a UV-vis spectrometer to determine the concentration of the DOX.In vitro cellular uptake
To observe cellular uptake of Fe3O4@DMSSs, MCF-7 cells were cultured for 12 h at 37 °C in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) in a 35 mm confocal dish and incubated for 24 h at 37 °C. After that, FITC-Fe3O4@DMSSs and DOX/Fe3O4@DMSSs were added into the dishes at a concentration of 100 μg mL−1, respectively. After incubation for 4 h, cells were washed for several times with PBS, fixed with 4% paraformaldehyde, and stained with 4′,6-diamidino-2-phenylindole (DAPI) for 5 min followed by washing with PBS. The fluorescence images were acquired by confocal laser scanning microscopy (CLSM).In vitro cytotoxicity
For the cytotoxicity of free DOX, parent Fe3O4@DMSSs and DOX/Fe3O4@DMSSs against MCF-7 cells and the biocompatibility of parent Fe3O4@DMSSs with L02 cells (normal human liver cells), cells were cultured in DMEM containing 10% FBS. Cells were seeded in 96-well plates at a density of 104 cells per well and cultured in 5% CO2 at 37 °C for 24 h. Then, free DOX, parent Fe3O4@DMSSs and DOX/Fe3O4@DMSSs were added to the culture medium, and the cells were incubated in 5% CO2 at 37 °C for 24 h. The concentrations of DOX were set at 0, 1.25, 2.5, 5, 10 and 20 μg mL−1, and the concentrations of parent Fe3O4@DMSSs were 0, 10, 25, 50, 100 and 200 μg mL−1. Cell viability was determined using the standard 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay. The statistical analysis of the experiments data utilized the Student's t-test. Each data point is represented as mean ± standard deviation (SD) of three independent experiments.Characterization
X-ray diffraction (XRD) patterns were obtained on Rigaku D/Max-2200PC using Cu Kα radiation (40 kV, 40 mA). Transmission electron microscopy (TEM) images were obtained on a JEM-2100F electron microscope operating at 200 kV. The samples were suspended in ethanol and then transferred onto a copper mesh coated with an amorphous carbon film for TEM measurements. Nitrogen adsorption–desorption isotherms at 77 K were measured on a Quantachrome NOVA 4200e. The specific surface area and the pore size distribution were calculated by using the Brunauer–Emmett–Teller (BET) and the Barrett–Joyner–Halenda (BJH) methods, respectively. Dynamic light scattering (DLS) measurements were performed using a Zeta potential/particle Sizer Nicomp TM 380 ZLS (PSS Nicomp particle size system, U.S.A.). UV-vis spectra were recorded on a Shimadzu UV-2550 spectrometer.Acknowledgements
This work was financially supported by the National Basic Research Program of China (973 Program, 2012CB933602); Program for New Century Excellent Talents in University (NCET-10-0379); NSFC (Grant nos 51172070, 51132009, 51202068, 5141101086); Shuguang Project (11SG30); Chenguang Project (13CG25); the Fundamental Research Funds for the Central Universities.Notes and references
- E. Katz and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 6042–6108 CrossRef CAS PubMed.
- R. Hao, R. J. Xing, Z. C. Xu, Y. L. Hou, S. Gao and S. H. Sun, Adv. Mater., 2010, 22, 2729–2742 CrossRef CAS PubMed.
- L. Y. T. Chou, K. Ming and W. C. W. Chan, Chem. Soc. Rev., 2011, 40, 233–245 RSC.
- Y. Hou, R. R. Qiao, F. Fang, X. X. Wang, C. Y. Dong, K. Liu, C. Y. Liu, Z. F. Liu, H. Lei, F. Wang and M. Y. Gao, ACS Nano, 2012, 7, 330–338 CrossRef PubMed.
- A. H. Lu, E. L. Salabas and F. Schuth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed.
- J. H. Gao, H. W. Gu and B. Xu, Acc. Chem. Res., 2009, 42, 1097–1107 CrossRef CAS PubMed.
- N. Lee and T. Hyeon, Chem. Soc. Rev., 2012, 41, 2575 RSC.
- K. Turcheniuk, A. V. Tarasevych, V. P. Kukhar, R. Boukherroub and S. Szunerits, Nanoscale, 2013, 5, 10729–10752 RSC.
- R. Y. Hong, B. Feng, L. L. Chen, G. H. Liu, H. Z. Li, Y. Zheng and D. G. Wei, Biochem. Eng. J., 2008, 42, 290–300 CrossRef CAS PubMed.
- S. Santra, R. Tapec, N. Theodoropoulou, J. Dobson, A. Hebard and W. H. Tan, Langmuir, 2001, 17, 2900–2906 CrossRef CAS.
- S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, J. Am. Chem. Soc., 2004, 126, 273–279 CrossRef CAS PubMed.
- J. Park, K. J. An, Y. S. Hwang, J. G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
- H. Deng, X. L. Li, Q. Peng, X. Wang, J. P. Chen and Y. D. Li, Angew. Chem., Int. Ed., 2005, 44, 2782–2785 CrossRef CAS PubMed.
- J. Kim, Y. Piao and T. Hyeon, Chem. Soc. Rev., 2009, 38, 372–390 RSC.
- J. Xie, C. Xu, N. Kohler, Y. Hou and S. Sun, Adv. Mater., 2007, 19, 3163–3166 CrossRef CAS PubMed.
- J. H. Lee, Y. M. Huh, Y. W. Jun, J. W. Seo, J. T. Jang, H. T. Song, S. Kim, E. J. Cho, H. G. Yoon, J. S. Suh and J. Cheon, Nat. Med., 2006, 13, 95–99 CrossRef PubMed.
- H. Ai, C. Flask, B. Weinberg, X. Shuai, M. D. Pagel, D. Farrell, J. Duerk and J. M. Gao, Adv. Mater., 2005, 17, 1949–1952 CrossRef CAS PubMed.
- D. Niu, Z. Zhang, S. Jiang, Z. Ma, X. Liu, Y. Li, L. Zhou, C. Liu, Y. Li and J. Shi, J. Mater. Chem., 2012, 22, 24936–24944 RSC.
- C. Zhang, B. Wangler, B. Morgenstern, H. Zentgraf, M. Eisenhut, H. Untenecker, R. Kruger, R. Huss, C. Seliger, W. Semmler and F. Kiessling, Langmuir, 2007, 23, 1427–1434 CrossRef CAS PubMed.
- C. Lu, Y. Hung, J. Hsiao, M. Yao, T. Chung, Y. Lin, S. Wu, S. Hsu, H. Liu, C. Mou, C. Yang, D. Huang and Y. Chen, Nano Lett., 2007, 7, 149–154 CrossRef CAS PubMed.
- I. I. Slowing, J. L. Vivero-Escoto, C. W. Wu and V. S. Y. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278–1288 CrossRef CAS PubMed.
- F. Q. Tang, L. L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed.
- S. H. Wu, C. Y. Mou and H. P. Lin, Chem. Soc. Rev., 2013, 42, 3862–3875 RSC.
- D. Niu, Z. Liu, Y. Li, X. Luo, J. Zhang, J. Gong and J. Shi, Adv. Mater., 2014, 26, 4947–4953 CrossRef CAS PubMed.
- J. Kim, H. S. Kim, N. Lee, T. Kim, H. Kim, T. Yu, I. C. Song, W. K. Moon and T. Hyeon, Angew. Chem., Int. Ed., 2008, 47, 8438–8441 CrossRef CAS PubMed.
- J. E. Lee, N. Lee, H. Kim, J. Kim, S. H. Choi, J. H. Kim, T. Kim, I. C. Song, S. P. Park, W. K. Moon and T. Hyeon, J. Am. Chem. Soc., 2009, 132, 552–557 CrossRef PubMed.
- M. Liong, J. Lu, M. Kovochich, T. Xia, S. G. Ruehm, A. E. Nel, F. Tamanoi and J. I. Zink, ACS Nano, 2008, 2, 889–896 CrossRef CAS PubMed.
- Z. Chen, D. Niu, Y. Li and J. Shi, RSC Adv., 2013, 3, 6767–6770 RSC.
- H. Wu, S. Zhang, J. Zhang, G. Liu, J. Shi, L. Zhang, X. Cui, M. Ruan, Q. He and W. Bu, Adv. Funct. Mater., 2011, 21, 1850–1862 CrossRef CAS PubMed.
- C. C. Huang, C. Y. Tsai, H. S. Sheu, K. Y. Chuang, C. H. Su, U. S. Jeng, F. Y. Cheng, C. H. Su, H. Y. Lei and C. S. Yeh, ACS Nano, 2011, 5, 3905–3916 CrossRef CAS PubMed.
- D. Niu, X. Liu, Y. Li, Z. Ma, W. Dong, S. Chang, W. Zhao, J. Gu, S. Zhang and J. Shi, J. Mater. Chem., 2011, 21, 13825–13831 RSC.
- C. Yang, W. Guo, L. Cui, N. An, T. Zhang, H. Lin and F. Qu, Langmuir, 2014, 30, 9819–9827 CrossRef CAS PubMed.
- S. Tan, Q. Wu, J. Wang, Y. Wang, X. Liu, K. Sui, X. Deng, H. Wang and M. Wu, Microporous Mesoporous Mater., 2011, 142, 601–608 CrossRef CAS PubMed.
- Z. Cheng, P. Ma, Z. Hou, W. Wang, Y. Dai, X. Zhai and J. Lin, Dalton Trans., 2012, 1481–1489 RSC.
- R. Li, L. Li, Y. Han, S. Gai, F. He and P. Yang, J. Mater. Chem. B, 2014, 2, 2127–2135 RSC.
- H. S. Yoo, E. A. Lee and T. G. Park, J. Controlled Release, 2002, 82, 17–27 CrossRef CAS.
- S. Ganta, H. Devalapally, A. Shahiwata and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS PubMed.
- Y. Kang and T. A. Taton, Angew. Chem., Int. Ed., 2005, 117, 413–416 CrossRef PubMed.
- D. Niu, Z. Ma, Y. Li and J. Shi, J. Am. Chem. Soc., 2010, 132, 15144–15147 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: For additional characterization. See DOI: 10.1039/c5ra04970a |
| This journal is © The Royal Society of Chemistry 2015 |