DOI:
10.1039/C5RA04081G
(Review Article)
RSC Adv., 2015,
5, 39726-39745
Labelling of brassinosteroids by isotopes of hydrogen and carbon
Received
7th March 2015
, Accepted 27th April 2015
First published on 30th April 2015
Abstract
The brassinosteroids (BRs) are a class of native plant growth regulating substances with high biological activity even at very low concentration. These compounds have been rigorously explored and it has been found that they are not only growth regulators in plants but also promising antiviral agents. Recently, it has been reported that natural BRs exhibit relatively interesting anticancer activities. Up to now, the basic anticancer potential of BRs against several normal and human cancer cell lines has been determined. Natural BRs, at micromolar concentrations, impart cell growth-inhibitory responses in several human cancer cell lines without affecting the normal cells. To study the mechanism of action of BRs at the molecular level, the corresponding isotopically labelled compounds are essential. The latter BRs are essential for the investigation of biosynthesis, metabolism, transport and distribution in plants. This venture ultimately led us to explore the labeling of BRs by isotopes of hydrogen and carbon and the related technique to do this. The present review will shed light on the synthetic avenues in this field from the time of the discovery of labelled BRs up until their most recent advances.
 Mahadeo R. Patil | Mahadeo R Patil received his MSc degree in Organic Chemistry in 2007 from S. R. T. M. University, Nanded, Maharashtra, India. Then he moved to NCL, Pune, India to work as a research assistant. Further, he spent 4 years at IOCB, Prague, Czech Republic to work on research project as an Assistant Scientist. There his research work focused on the synthesis of isotopically labelled brassinosteroids and their application. Presently he is a Doctoral student in CNMS, Bangalore, India working with Dr. Rangappa Keri and his research is mainly focused on synthesis and biological activity of novel heterocyclic compounds. |
 Tomáš Elbert | Tomaš Elbert earned his Ph.D. degree working on sugar chemistry at the Faculty of Natural Sciences, Charles University, Prague, in 1980. He started to work on radioisotopes in 1980 at the Institute for Research, Production and Application of Radioisotopes, Prague. After that he served as Assistant professor of Organic Chemistry at Faculty of Natural Sciences, Charles University. Presently, he is head of the Laboratory of Radioisotopes at the Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences. In 2011 he was awarded with the IIS-CED Award 2011 for his work in the Advisory Board of IIS-CED. |
 Rangappa S. Keri | Dr. Rangappa Keri is currently working as an Assistant Professor at Jain University, Bangalore, Karnataka, India. Before joining Jain University, he did his postdoctoral work at IST, Universidade de Lisboa, Lisboa, Portugal and Kyung Hee University, Seoul, South Korea. His research interests concern Organic and Medicinal chemistry. Especially, development of new drugs including acetyl acetylcholinesterase (AChE) inhibitors, anti-cancer, neuroprotective agents and anti-virals by organic synthesis. Also, development of organometal and metaloorgano-catalyzed enantioselective methods for the synthesis of heterocyclic scaffolds. |
1. Introduction
Brassinosteroids (BRs) represent a class of naturally occurring phytohormones with various physiological activities and ubiquitous distribution in the plant kingdom.1 The vicinal diol grouping on ring A is typical of the BR-plant hormones discovered thirty years ago.2 In Fig. 1, the formulae of two typical BRs-24-epibrassinolide 1 and castasterone 2 – are given. These compounds have been extensively studied and it has been found that they exhibit not only growth regulation functions in plants but also promising antiviral activity.3
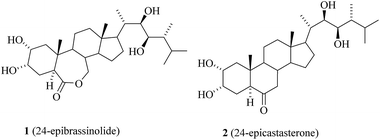 |
| | Fig. 1 Structures of 24-epibrassinolide (24-epiBL) and 24-epicastasterone (24-epiCS). | |
Recently, molecular studies directed towards the essential role of BRs in plant growth and development4 and their chemical synthesis, biological mode of action, and practical application in agriculture and horticulture5,6 have greatly intensified. The essentialness of isotopically labelled BRs in the investigation of the biosynthesis, metabolism, transport and distribution of endogenous BRs in plants has been documented. In such studies, BRs labelled with isotopes of hydrogen are most frequently used to explain the biosynthesis, metabolism and mode of action at a molecular level.
Therefore, the numerous strategies for the synthesis of BRs labelled with deuterium (2H or D) and tritium (3H or T) in the side chain or in the ring system have been developed.7 BRs are formed by a biosynthetic network of alternative pathways and sub-pathways. A number of feeding experiments using labelled BRs precursors as a substrate are necessary to elucidate these pathways. In these reactions, labelled BRs are being used as internal standards for qualitative analysis of endogenous BRs and they have been employed in biosynthetic experiments.8–11 Some of the deuterium-labelled 24-methylenecholesterol and related C28 steroids were used as both, internal standards in quantitative analyses and substrate, for metabolic studies in BRs deficient mutants of Arabidopsis thaliana and Pisumsativum. The biosynthetic sequence from campesterol12 to campestanol in Arabidopsis thaliana was determined by identification of each intermediate followed by feeding experiments with deuterium-labelled intermediates.13,14 The dwarf pea (Pisumsativum) mutants Ika and Ikb are BRs insensitive and deficient, respectively. The latter mutant was rescued to wild type by exogenous application of labelled brassinolide and its precursors. Feeding experiments using deuterium labelled 24-methylenecholesterol indicated that an Ikb mutant is unable to isomerize and/or reduce the double bond.
Recently, brassinolide biosynthetic pathways have been elucidated by feeding deuterium-labelled intermediate to suspension cultures of Catharanthus roseus.15,16 Identification of cathasterone with one of the hydroxyl groups lacking and application of its labelled analogue in feeding experiments9 demonstrated that the brassinolide biosynthesis proceeds via initial hydroxylation at C-22 followed by an introduction of the hydroxyl group at C-23. Deuterium labelled of secasterol, teasterone and typhasterol, upon administration to rye seedlings, were incorporated into secasterone and 2,3-diepisecasterone, indicating a biosynthetic route via teasterone/typhasterol to secasterol to 2,3-epoxybrasinosteroids, secasterone in seedlings of Secale cereal. Similarly, deuterated secasterone upon administration resulted in deuterated castasterone and 2-epicastasterone used in biosynthetic sub-pathways from typhasterol/teasterone via 2,3-epoxybrassinosteroids intermediate to castasterone (CS).17 Some of the 14C labelled BRs, namely (22R,23R)- and (22S,23S)-[4-14C]-24-epiBL (A and B in Scheme 11, respectively) were used to facilitate metabolic and distribution studies of (22R,23R)-24-epiBL and (22S,23S)-24-epiBL as well as to study their role in the growth of grain and vegetables as they are promising candidates for agriculture application.18 Predominantly the 14C-labelled epiBL (Scheme 10) was used in the uptake and the transport study of exogenously applied epibrassinolide on seedlings of cucumber and wheat. When applied to roots, 14C-epiBL was readily taken up and swiftly transported throughout both the plant species. When 14C-epiBL was applied to the adaxial surface of a young cucumber leaf, it was readily taken up, however, transported very slowly compared with the previous case. In wheat leaves, 14C-epiBL was transported only in the apical direction from the treated spot after 3 days of treatment; however, it was not transported from the treated leaf to the other leaves or organs even after seven days. Recently, isotopically labelled [7,7-2H2] epibrassinolide was used for biosynthetic transformation studies.19 This compound found useful for biochemical and physiological investigation in the plant.20 Simultaneously, it was also discovered that natural BRs exhibit relatively interesting anticancer activities. So far, potential anticancer activities of 24-epiBL 1 and 24-epicastasterone 2 on several human cancer cell lines have been determined.19 24-Episecasterol was prepared and found cytotoxic against human breast carcinoma MCF-7 (Michigan Cancer Foundation) cells.21 It was also demonstrated that non-plant cells, yeast WAT21, generates a steroidal plant hormone castasterone. To understand how castasterone is generated in WAT21 cells, deuterium labelled 6-deoxo-[26,28-2H6]teasterone(TE), 6-deoxo-[26,28-2H6]typhasterol (TY) and 6-deoxo-[26,28-2H6](CS) were fed to WAT21 cells and their metabolites were isolated. [26,27-2H6] labelling of brassinolide, castasterone, typhasterol, and teasterone were reported by Takatsuto and Ikekawa in 1986.22 Deuterium labelled 6-deoxo-BRs were identified as biosynthetic precursors of CS in WAT21 cells.23 In this review, a variety of labelling procedures of BRs with stable and radioactive isotopes of hydrogen and carbon will be presented.
2. Synthesis of deuterium-labelled BRs
2.1. BRs with deuterium labels in the side chain
2.1.1. [26-2H3], [26,27-2H6] and [26,28-2H6] labelled BRs.
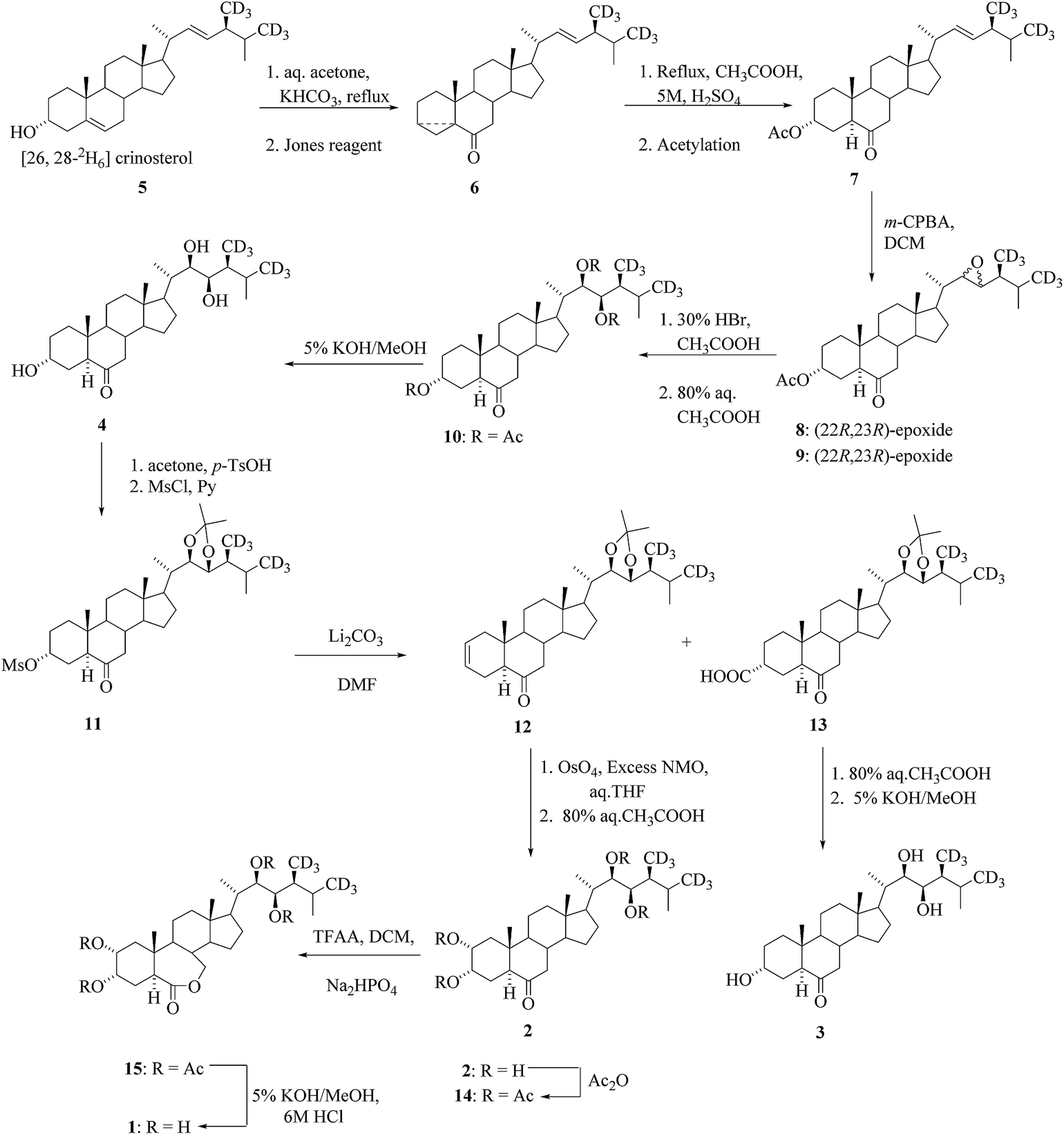
Brassinolide (BL) and related C-28 BRs as castasterone (CS), typhasterol (TY), and teasterone (TE) occur in a wide variety of higher plants.24,25 The synthesis of [26,28-2H6] brassinolide 1, [26,28-2H6]CS 2, [26,28-2H6]TY 3 and [26,28-2H6]TE 4 as an internal standards for GC-MS assays of BRs was reported in the literature.26,27 [26,28-2H6]crinosterol285 served as a starting material for the synthesis of four deuterated BRs (1–4) (Scheme 1).
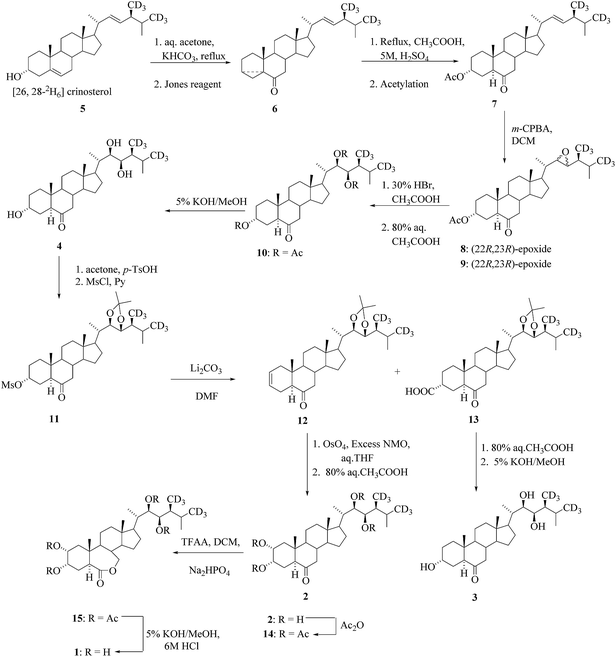 |
| | Scheme 1 Synthesis of [26,28-2H6]BL (1), [26,28-2H6]CS (2), [26,28-2H6]TY (3) and [26,28-2H6]TE (4). | |
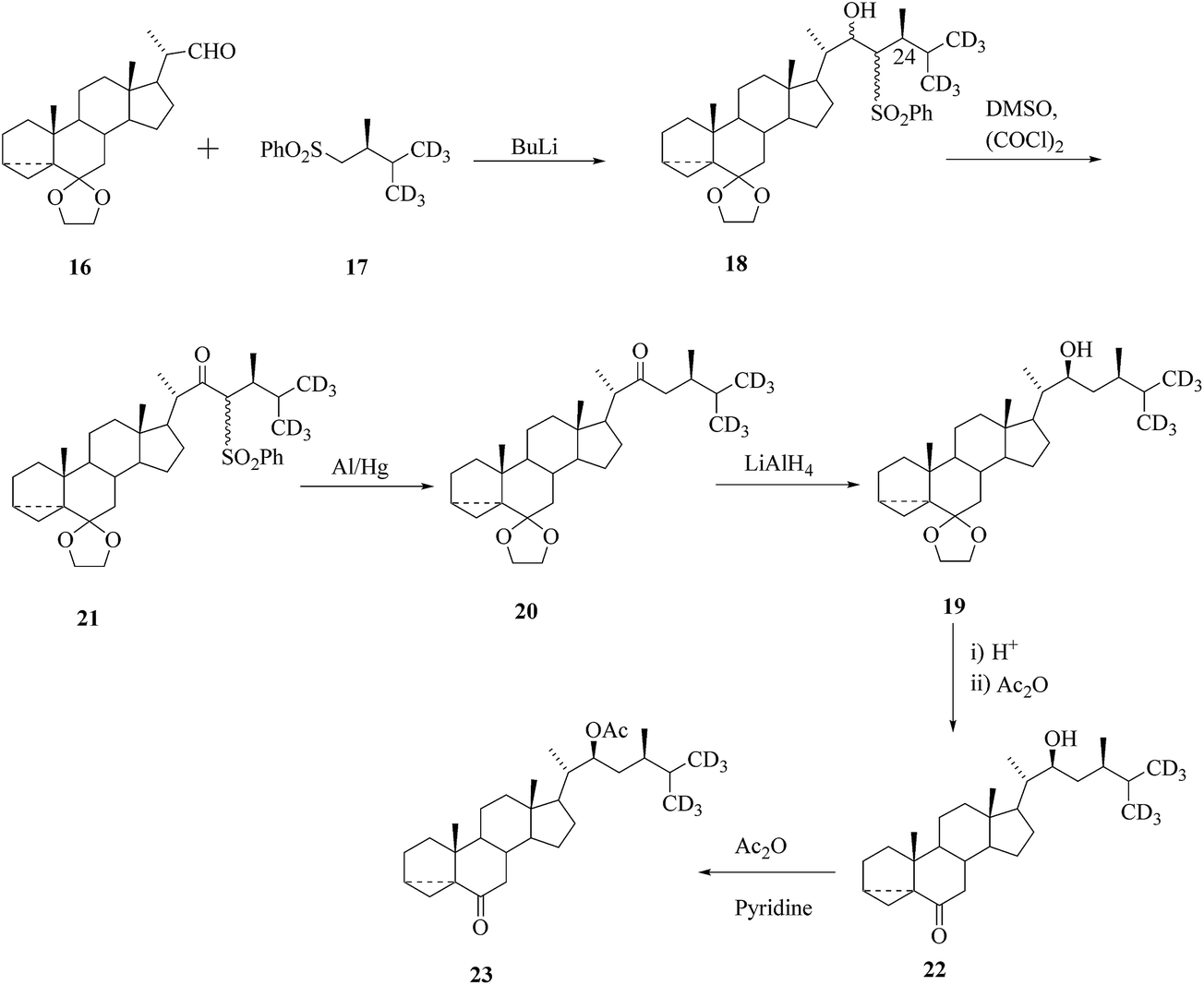
Alternatively, labelled BRs containing three or six deuterium atoms appended in the terminal methyl groups of the side chain (in a position ensuring lack of isotopic exchange) were prepared from stigmasterol or bisnorcholenic acid. There are main strategies for the construction of the side chain containing an asymmetric centre at C-24 of the compound 18 and these are based on the coupling of 16-aldehyde with an appropriate chiral sulfone synthon 17. These methodologies were also used for the stereoselective construction of the side chain labelled BRs. Preparation of trideuterated fragment 22 in the side chain of BRs29,30 through Claisen rearrangement for stereoselective construction of the steroidal side chain. Construction of the side chain of hexa-deuterated BR was performed via coupling of sulfone 17 with aldehyde 16 (Scheme 2).
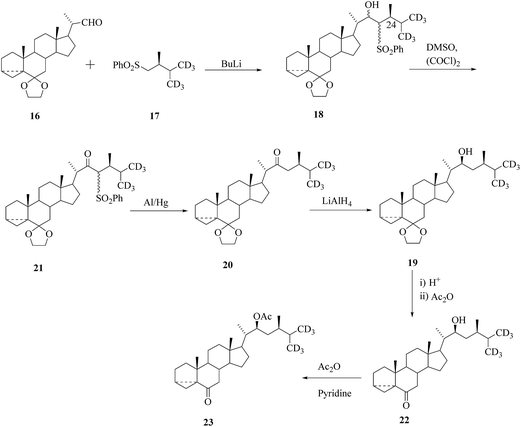 |
| | Scheme 2 Construction of the side chain of hexa-deuterated BRs. | |
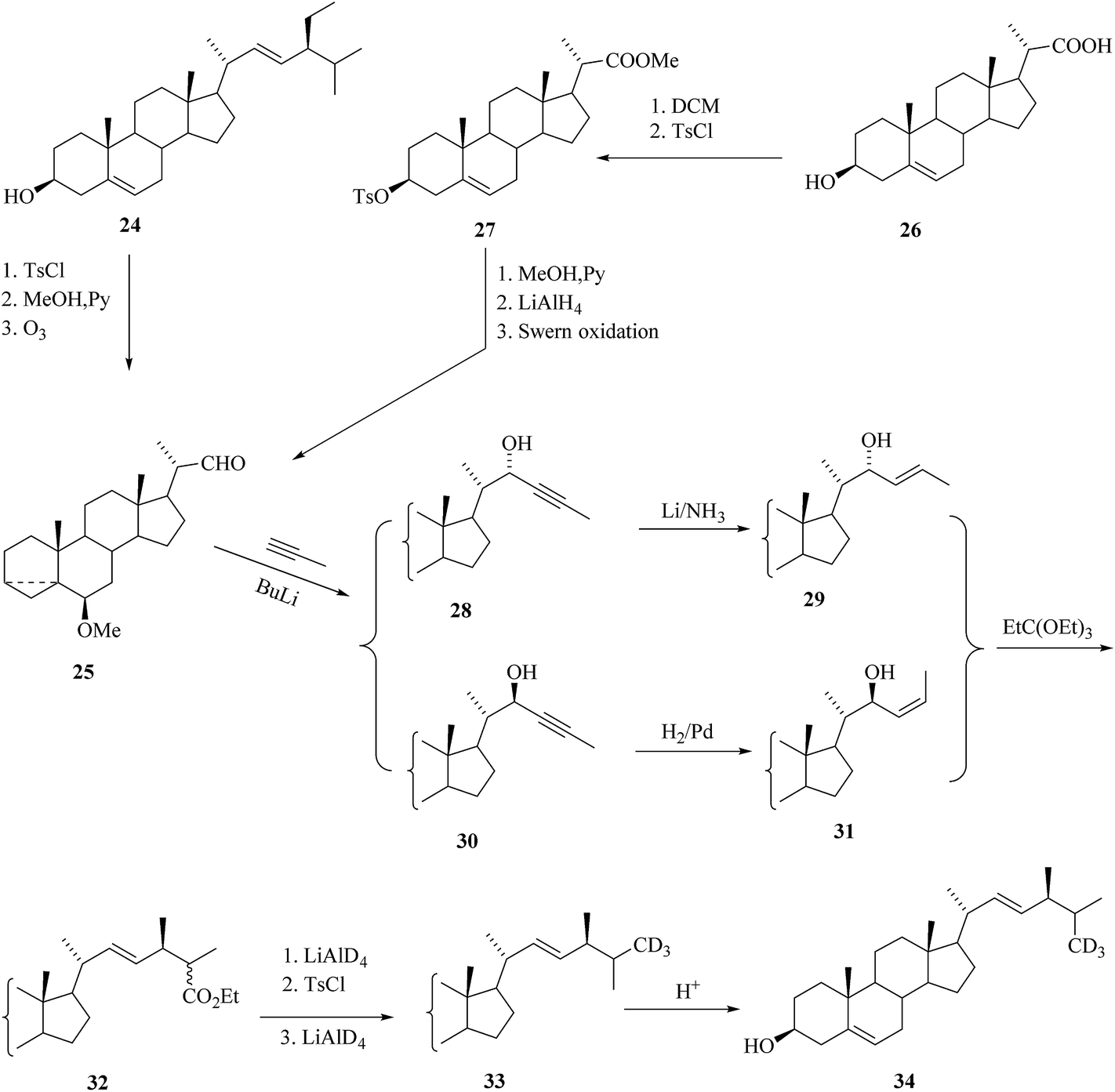
Introduction of functional groups at the cyclic part of the steroids could be done prior to the construction stages of the side chain based on various 22-aldehydes.31,32 As a result, the aldehyde 25 was the best choice for the synthesis of labelled BR derivatives with functional groups tethered to the cyclic part and it was suitable for the preparation of many highly functionalized BRs. The aldehyde 25 was prepared either from stigmasterol 24 by traditional method33,34 in 3 steps or from 23,24-bisnorcholenic acid3526 (Scheme 3). An important part of the convergent synthesis was the preparation of the chiral intermediate 17, which was accompanied through (2R)-3-hydroxy-2-methylpropanoate.35,36 The synthesis of precursor of [26-2H3]-brassinosteroids 34 for biochemical studies from the starting material 25 is depicted in Scheme 3. In the first convergent strategies based on the Claisen rearrangement of a single isomer of allylic alcohol is considered useful in this rearrangement to access certain product.27,28,37,38 A Claisen rearrangement has been widely used in the preparation of Δ22-steroids containing an alkyl substituent with predictable stereochemistry at C-24. Then the ester compounds 32 were prepared via isomeric acetylenic alcohols 28 and 30 and the allylic alcohol intermediates 29 and 31 (Scheme 3). Successive reduction of the ester 32 with LiAlD4 followed by tosylation and deuteride reduction gave compound 33 containing three deuterium atoms in the terminal part of the side chain.30 Regeneration of the cyclic part of 33 by acid treatment to yield the deuterated crinosterol 34 (Scheme 3).
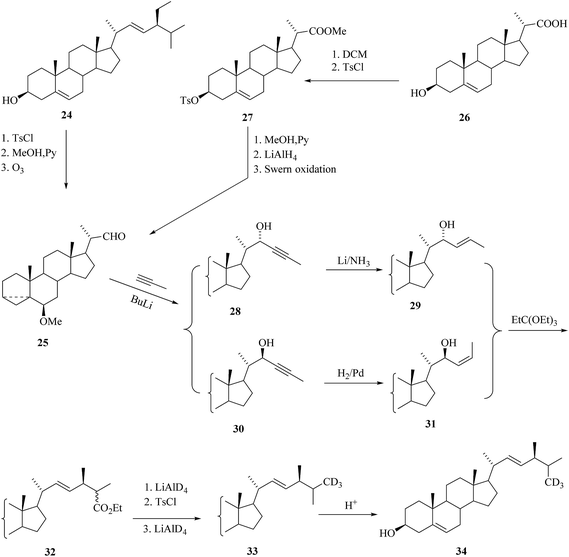 |
| | Scheme 3 Construction of the side chain of [26-2H3]BRs. | |
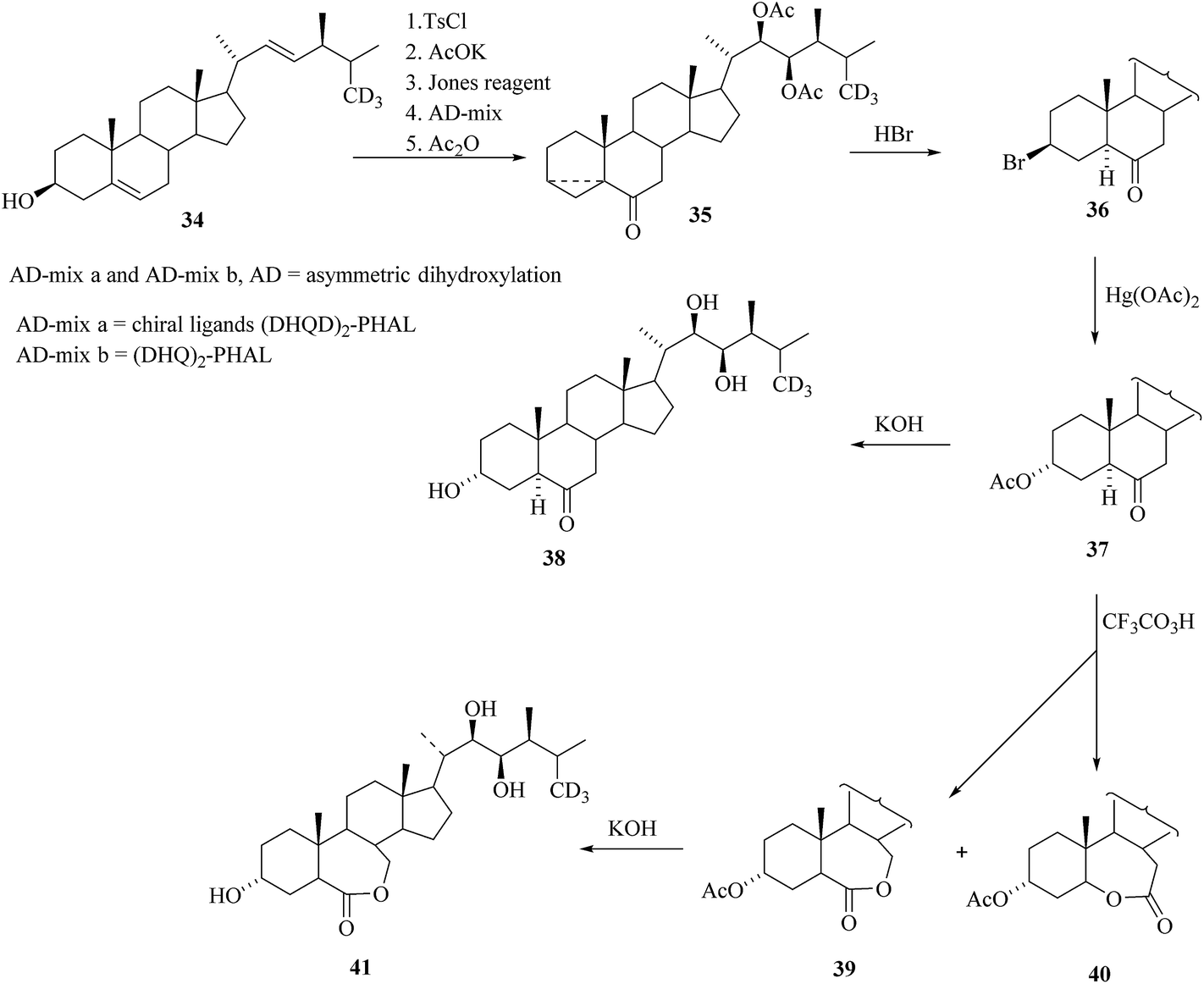
After formation of a 3-membered ring and oxidation on C-6 by standard sequence of reactions starting from the 34 and then the sharpless oxidation of side chain double bond using (DHQD)2-PHAL as a ligand and osmium tetraoxide as catalyst led to expected alcohol and followed by acetylation to obtained the compound 35 and then to bromide 36. Nucleophilic substitution of the bromide in 36 offered 3α-acetoxyketone 37 and after deacetylation [26-2H3]TY 38 was obtained. Baeyer–Villiger oxidation of 37 yielded lactone 39 along with its regioisomer 40. Deprotection of the hydroxyl groups furnished 2-dehydroxy-[26-2H3] brassinolide 41 (Scheme 4).
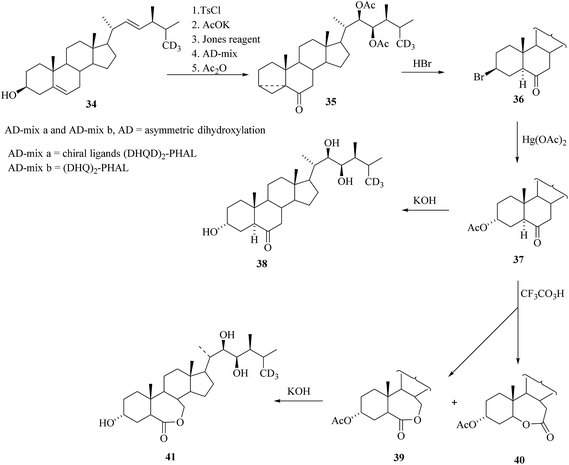 |
| | Scheme 4 Synthesis of [26-2H3]TY 38 and related compounds. | |
Bromide 36 found useful as an intermediate for the preparation of deuterated BRs with an epoxy group in ring A (Scheme 5). Dehydrobromination of 36 offered 6-ketone 42 as the main product of the reaction with formic ester 43 as a byproduct. Epoxidation of 42 ensued stereoselectively to yield 2α,3α-epoxyacetate 44 that was then deprotected to 2,3-[26-2H3]episecasterone 47. Compound 2,3-[26-2H3] secasterone 48 was obtained via bromohydrin 45 from 42. Formate 43 can be used for the preparation of 3,6-diketo BRs 49via 3-dehydroteasterone 46 (Scheme 5).
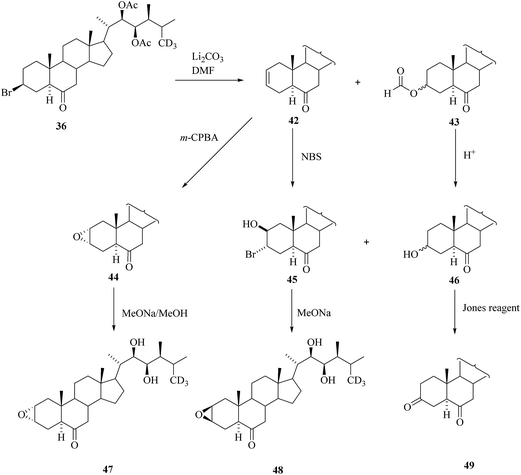 |
| | Scheme 5 Synthesis of deuterated BRs with an epoxy group in ring A. | |
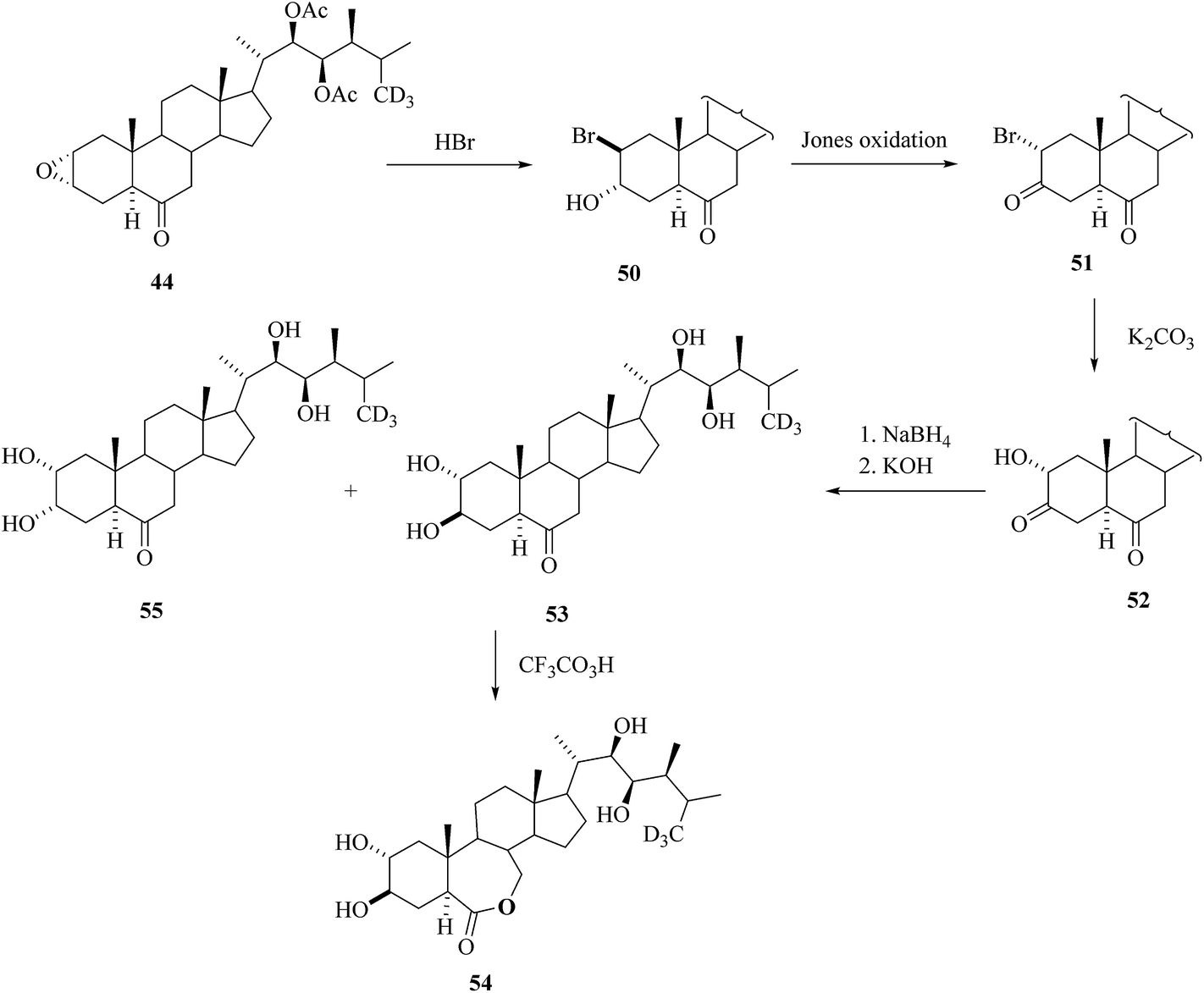
The epoxide 44 was used as key intermediate for the synthesis of [26-2H3] BRs 53–55 as depicted in Scheme 6.30trans-Diaxial ring opening of epoxide 44 with the action of HBr led to bromohydrin 50. Jones oxidation ensued with the formation of a ketonic group at C-3 and inversion of configuration at C-2 (bromoketone 51). After nucleophilic substitution of bromine by a hydroxyl group, the hydroxyl ketone 52 was isolated. The regiospecific and stereoselective reduction of 52 followed by saponification to remove acetate groups in the side chain furnished a mixture of 3-epi[26-2H3]CS 53 and [26-2H3]CS 55. Bayer–Villiger oxidation of 53 with trifluroperoxoacetic acid offered 3-[26-2H3]epiBL 54.
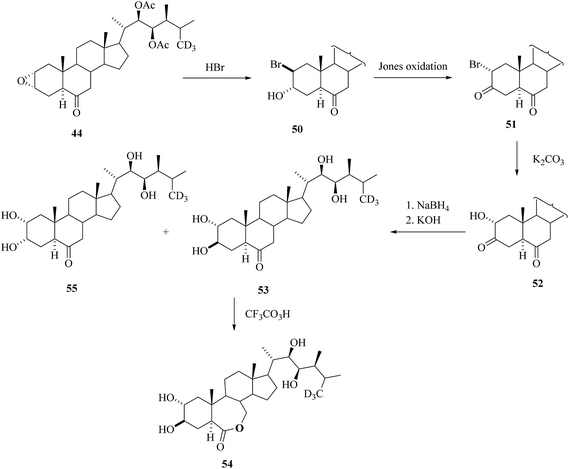 |
| | Scheme 6 Synthesis of deuterated tetra-hydroxyl BRs. | |
An important convergent approach using the side chain fragment was used for the preparation of the labelled BRs containing six deuterium atoms at C-26 and C-27.35,36 Coupling of the appropriate aldehyde with lithium salt of chiral sulfone led to compounds 56 (Scheme 7).
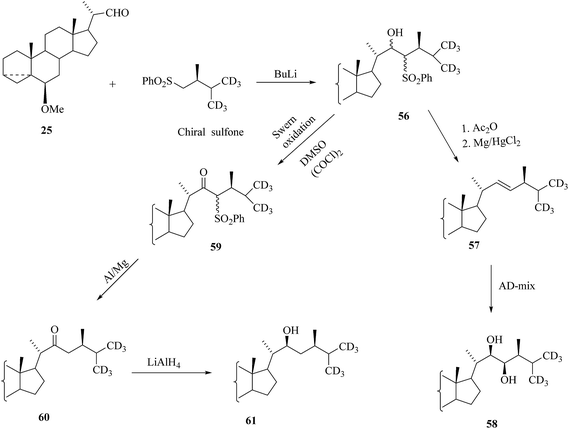 |
| | Scheme 7 Construction of the side chain of [26,27-2H6]BRs. | |
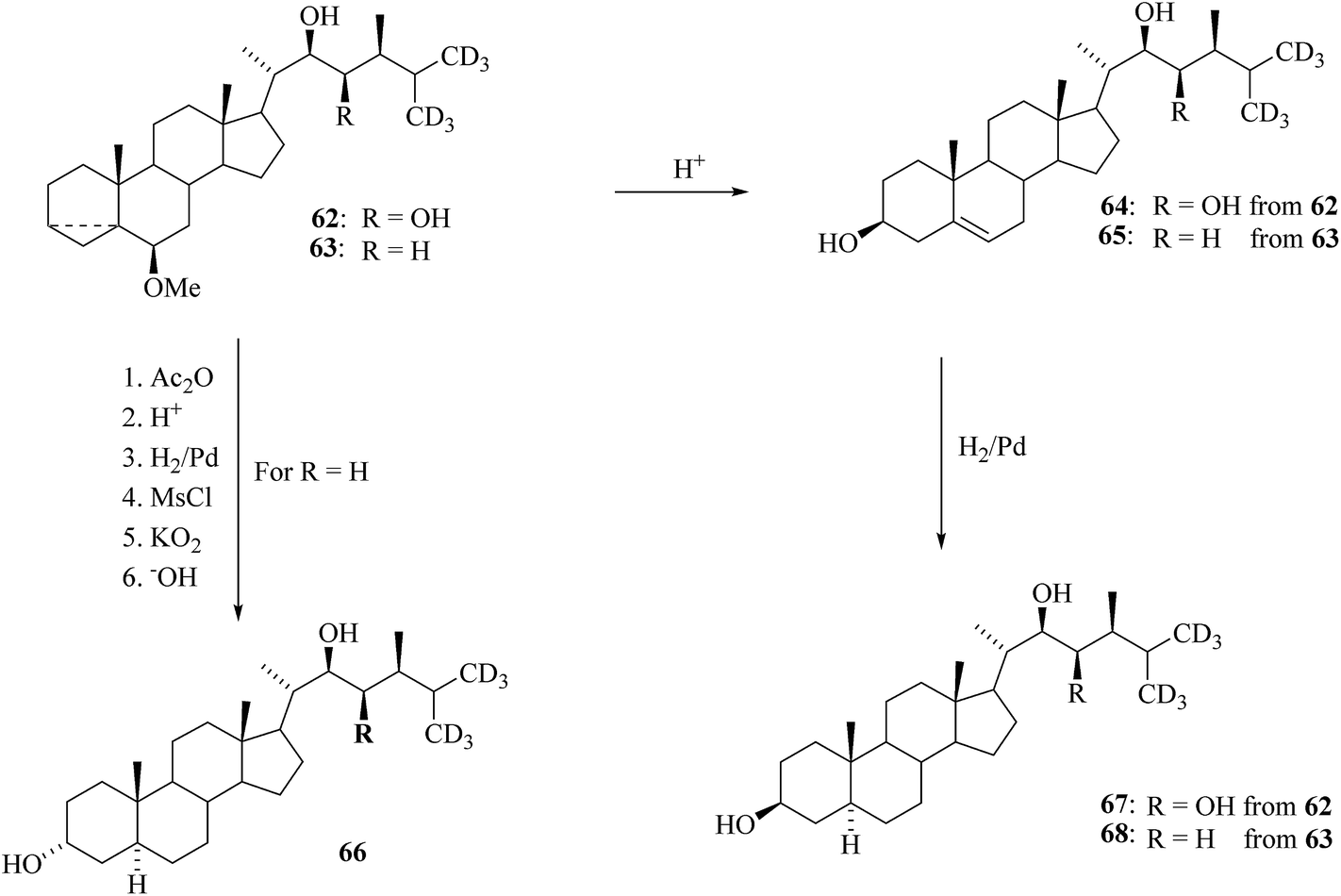
Then alcohol 56 is functionalized by modified Julia olefination protocol via acetylation followed by reduction elimination with magnesium amalgam to furnish the olefin 57. Introduction of a (22R,23R)-diol function was attained by using asymmetric dihydroxylation (AD) as discussed in the earlier case for [26-2H3] BRs. Synthesis of a derivative 61 demonstrated the methodology applied in the preparation of BRs containing one hydroxyl group in the side chain. Swern oxidation of hydroxyl sulfones 56 directed to the ketosulfones 59 and then desulfurization with aluminium amalgam was smoothly resulted in the formation of the ketones 60. The further reduction of 60 with lithium aluminium hydride to offered desired (22S)-alcohols 61 as the main product (Scheme 7). Mono- and dihydroxy derivatives 62 and 63, respectively allowed easy access to a variety of corresponding [26,27-2H6]BRs as shown in Scheme 8. Δ5-3β Alcohols 64 and 65 were synthesized from 62 and 63 in a dioxane–water solution olfin presence of toluenesulfonic acid at elevated temperature. Hydrogenation of double bond over palladium bequeathed 6-deoxoteasterone 67 and 6-deoxocathasterone 68. The preparation of 3α-alcohol 66 involved inversion configuration at C-3 via nucleophilic substitution of intermediate mesylate with potassium superoxide.
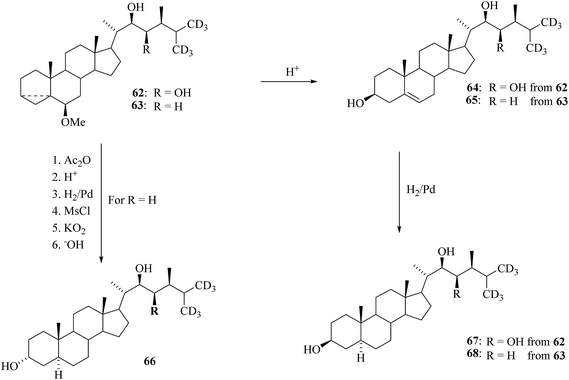 |
| | Scheme 8 Modification of rings A and B of [26,27-2H6]BRs. | |
An attempt to prepare labelled brassinolide biosynthetic precursors having 3,6-diketone moiety was first reported with by-products similar to 42 obtained from dehydrobromination of 3β-bromides (see also Scheme 5). It is proved incompatible with the removal of the acetate protecting groups in the side chain and the problem was solved by using aldehyde 69 as a key intermediate (Scheme 9). Essentially, the same protocol as that described in Scheme 7 was used for the preparation of 3-dehydro-[26,27-2H6]cathasterone 72 and 3-dehydro-[26,27-2H6] TE 73.35
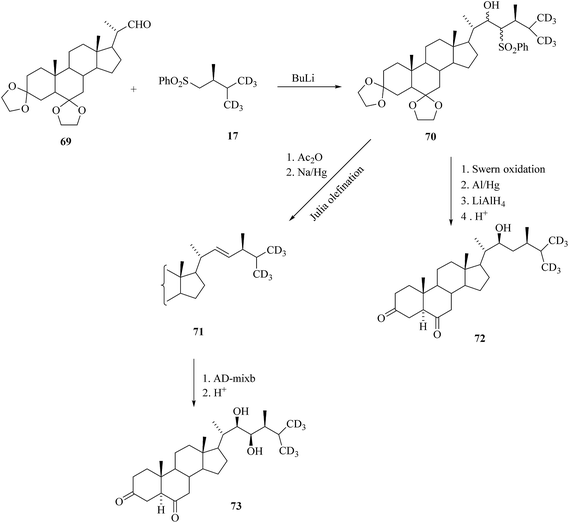 |
| | Scheme 9 Synthesis of 3,6-diketo-[26,27-2H6] BRs. | |
2.1.2. Synthesis of deuterio-labelled 24-methylenecholesterol and related steroids.
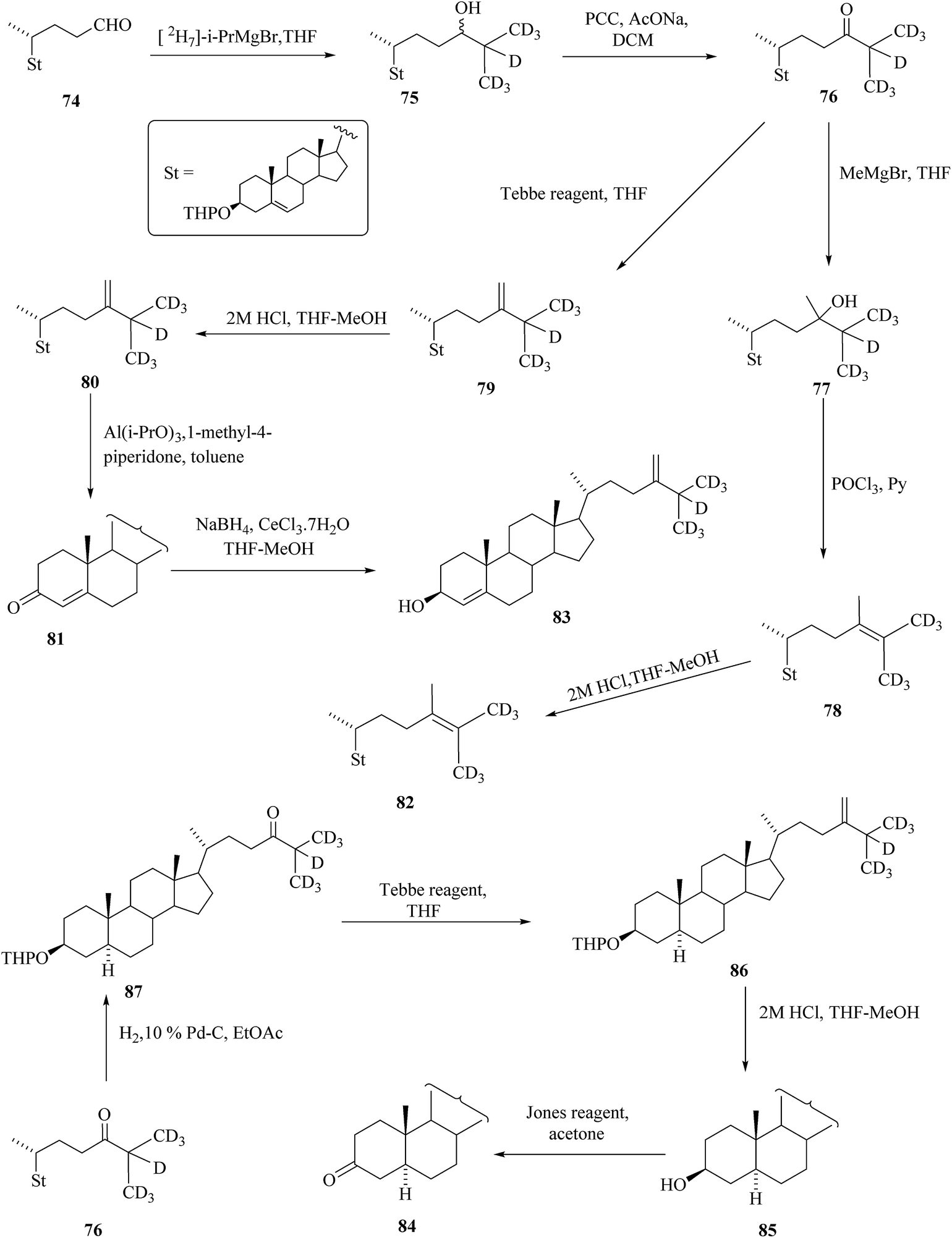
Anastasia et al. reported the synthesis of BRs with one deuterium atom,39 however, usually four and more deuterium atoms in the molecule are required for the MS standards. Contemporarily, Takatsuto et al. reported labelling of the side chain of targeted steroid by more than one deuterium atom. It is described in the work that the synthesis of deuterium labelled C28 steroids 80 to 86 from the known 3β-tetrahydropyranyloxychol-5-en-24-al 74 (Scheme 10).40
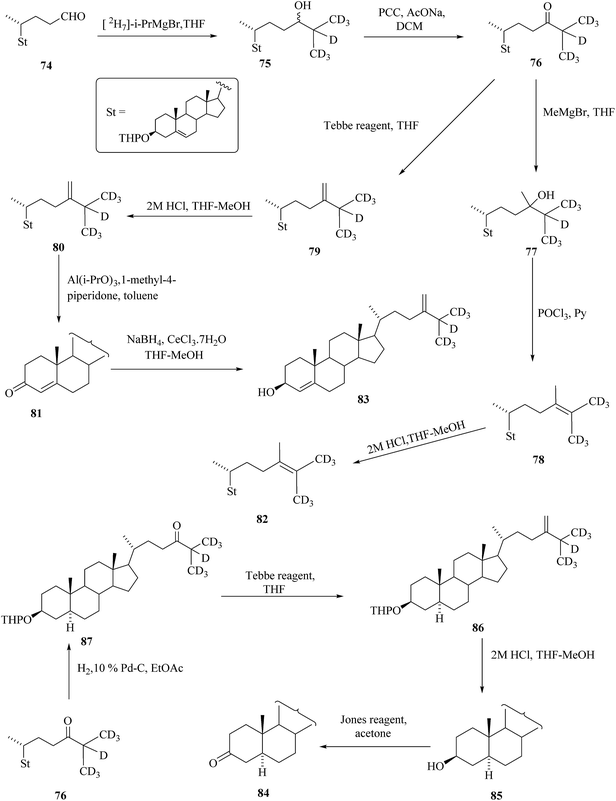 |
| | Scheme 10 Phytosterol and their deuterium-labelled analogues. | |
2.1.3. Synthesis of [25,26,27-2H7]BL from parent brassinolide.
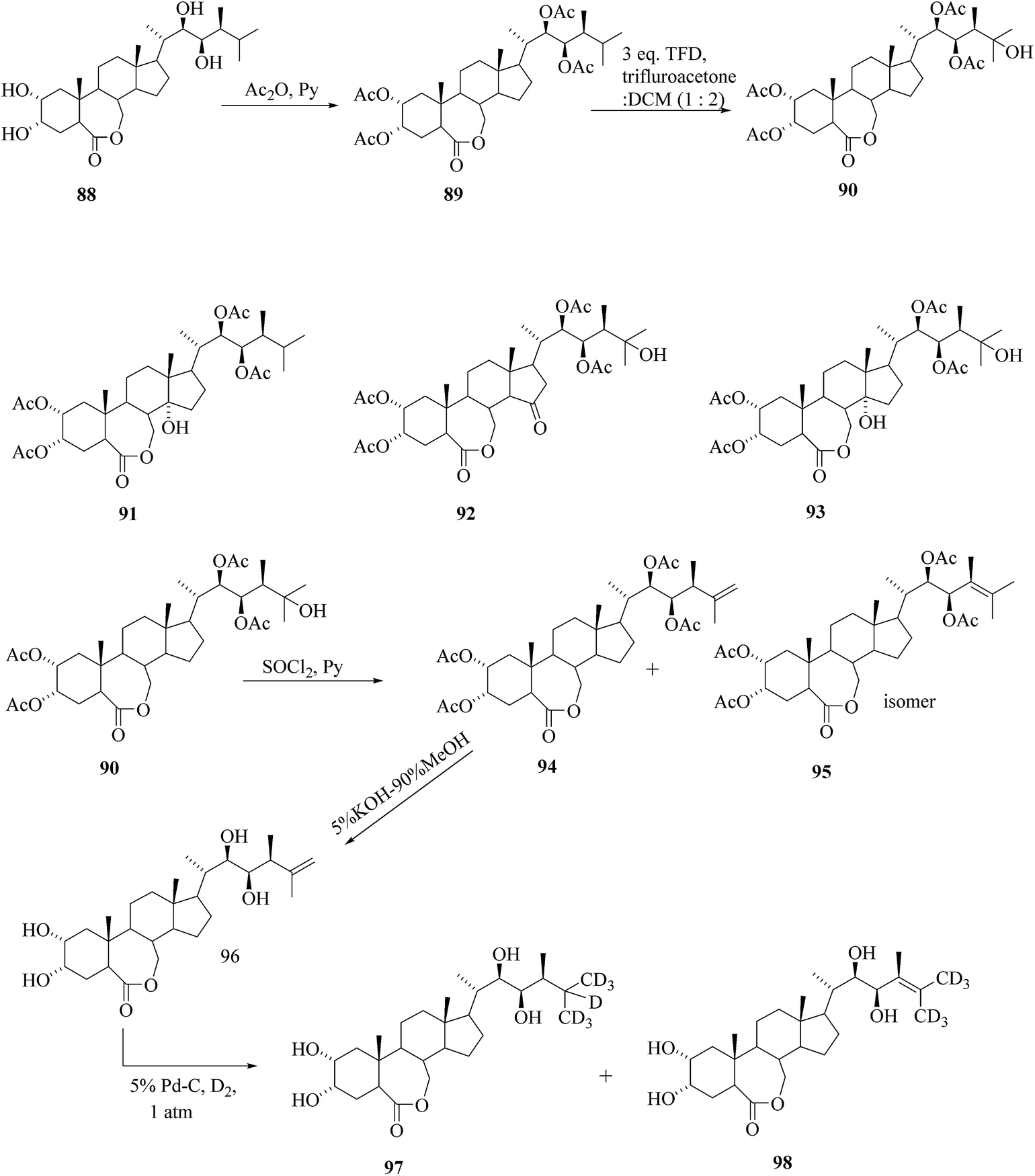
Several routes for the synthesis of labelled BRs have been reported so far,41 however, all are quite lengthy multi step reactions which are neither eco-friendly nor cost-effective. The easy way to synthesize the side chain labelled BRs from parent BRs includes three basic steps; C-25 hydroxylation, dehydration to create double bond between C25–C26 and deuteriogenation or tritiation of the double bond. Based on these basic steps, the preparation of [25,26,27-2H7]BL 97 from brassinolide 88 as a starting material was achieved42 (Scheme 11). 25-Oxy-functionalization of tetra-O-acetyl BL 89 with TFD offered predominantly 25-hydroxy compound 90 while 14-hydroxy, 25-hydroxy-15-oxo- and 14,25-dihydroxy derivatives 91, 92, 93, respectively were identified as byproducts. Dehydration of 90 headed to the formation of a mixture of the compounds 94 and 95 (which were unseparable) and then the mixture of isomers were subjected to deprotection of tetra-O-acetyl groups. Subsequently the mixture of isomers were treated with KOH in aqueous MeOH at room temperature until the lactone ring completely opened to the carboxylate and then at refluxing temperature. After recyclization of lactone ring and desalting the crystalline compound 96 was obtained. Reduction of the double bond of Δ25(26) BL 96 using deuterium gas and 5% Pd/C as catalyst was preceded with extensive isotopic exchange, which resulted in high deuterium incorporation leading to the formation of [25,26,27-2H7]BL 97 with 60% isotopic purity. The partial migration of double bond provided the minor byproduct Δ24(25) [26,27-2H6]BL 98 with 68% isotopic purity.
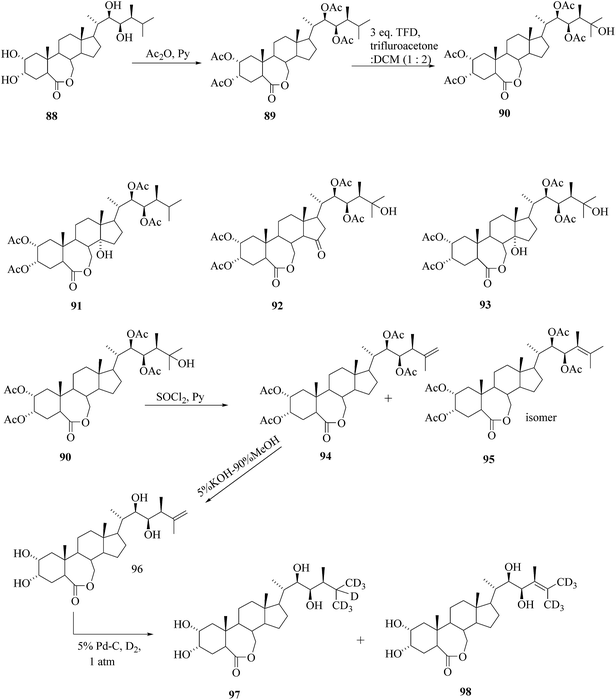 |
| | Scheme 11 Synthesis of [25,26,27-2H7]BL. | |
2.1.4. Alternative way to [26,28-2H6]brassinolide and [26,28-2H6]castasterone.
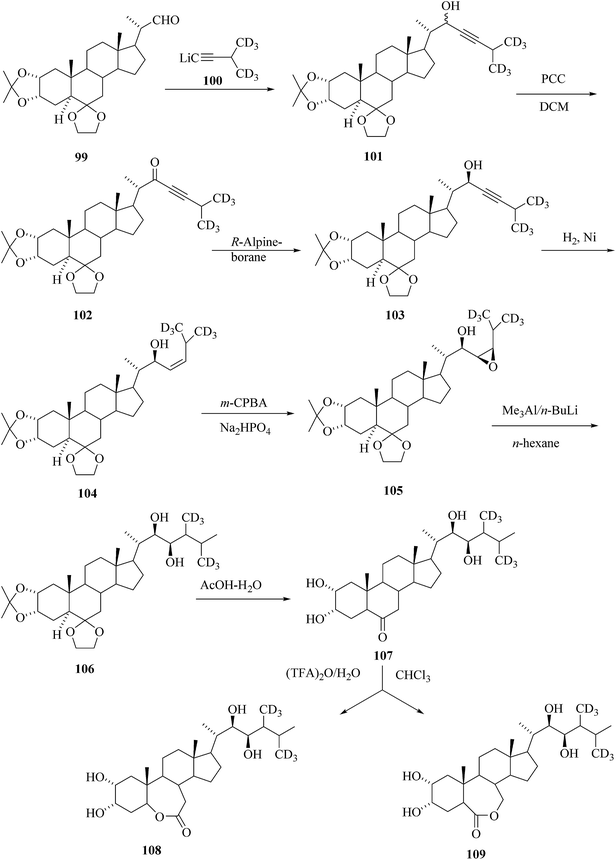
Kolbe et al.43 reported the synthesis of deuterium labelled BRs [26,28-2H6]CS 107 and [26,28-2H6]BL 108 starting from 6,6-ethylenedioxy-20-formyl-2α,3α-isopropylidenedioxy-5α-pregnane 99 and 3-[methyl-2H3]methyl-[4,4,4-2H3]but-1-yne 100, respectively (Scheme 12). The acetylenic alcohol 101 was oxidized by pyridinium chlorochromate and stereoselective reduction of ketone 102 yielded stereospecifically the acetylenic alcohol 103. Partial hydrogenation on Raney® Ni gave an allylic alcohol 104. The double bond was epoxidized by 3-chloroperbenzoic acid and then epoxide 105 was treated by trimethylaluminium/n-butyllithium reagent. Opening of the epoxide ring in 105 leads to the unusual methyl group migration from C-26 to C-25 took place during methylation and then subjected to hydrolysis to furnish [26,28-2H6]CS 107 was obtained instead of [26,27-2H6]-labelled isomer. Baeyer–Villiger oxidation of [26,28-2H6]castasterone 107 gave mixture of regioisomers 109 and 108 in which [26,28-2H6]BL 109 prevailed (109![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :108 = 6:1).
:108 = 6:1).
 |
| | Scheme 12 Synthesis of [26,28-2H6]CS and [26,28-2H6]BL. | |
2.1.5. Synthesis of [26-2H3]-epibrassinolide and its precursors.
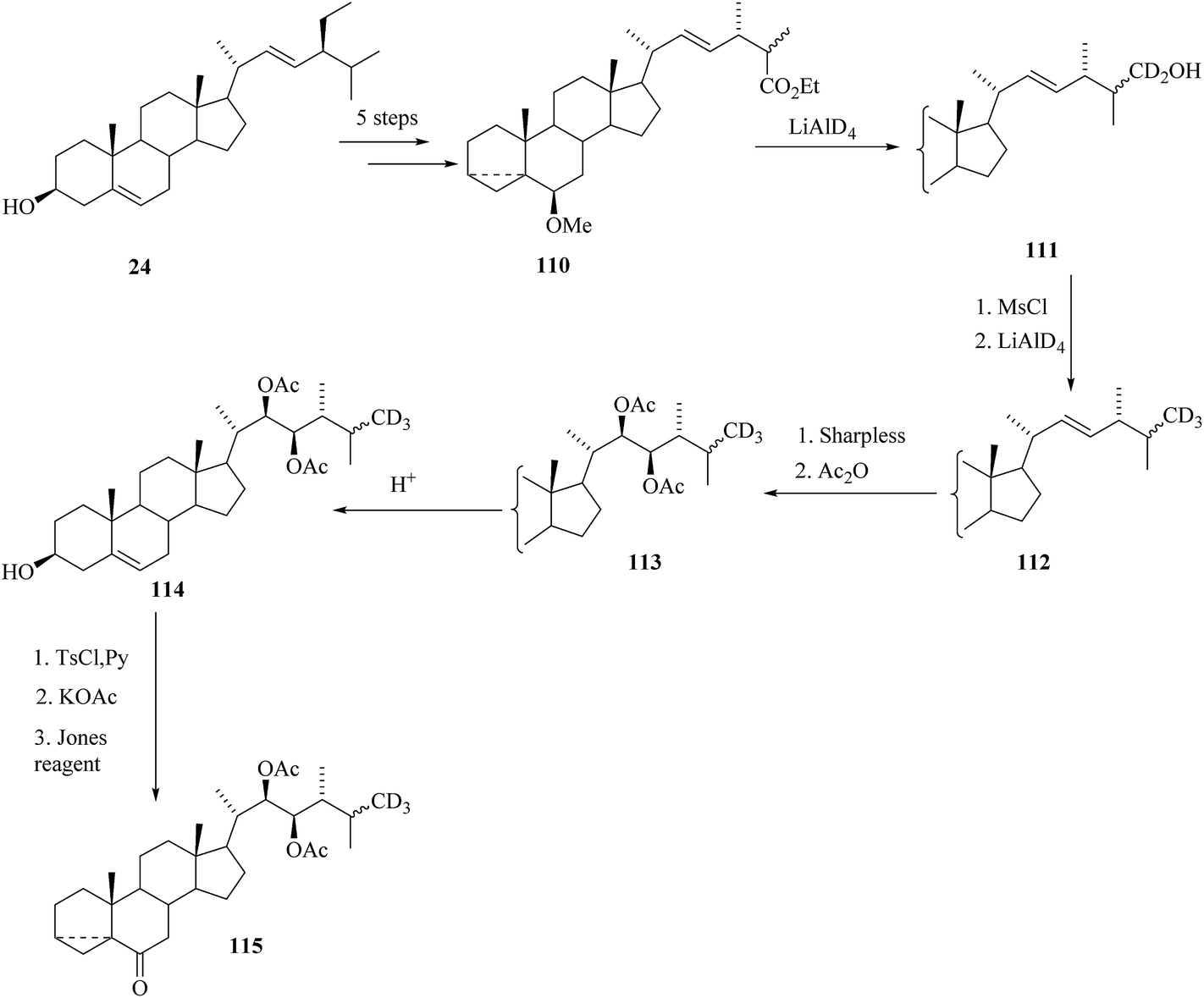
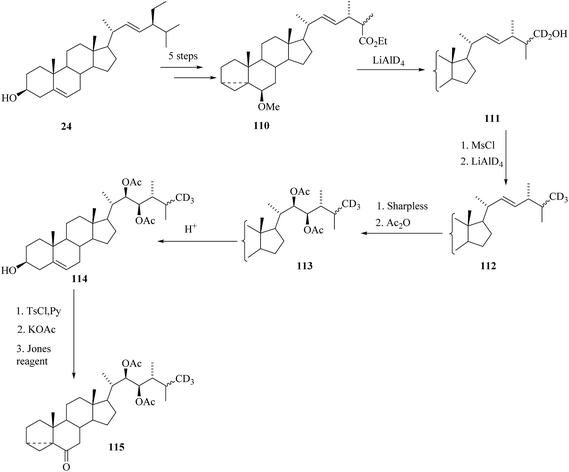
A further study towards the synthesis of labelled BRs containing three deuterium atoms in the terminal part of the side chain starting from commercially available stigmasterol 24 is reported by Khripach et al.32 3,5-cyclo ester 110 was prepared from stigmasterol 24 in 5 steps. Labelling with deuterium was done in two steps. Reduction of 3,5-cyclo ester 110 with lithium aluminium deuteride gave alcohol 111 (Scheme 13) and then mesylation of the alcohol 111 followed by reduction with LiAlD4 was introduced the third deuterium to the side chain end methyl group and thus Δ22-derivative 112 was obtained. Asymmetric dihydroxylation of the double bond in 112 followed by acetylation yielded (22R,23R)-diacetate 113. A methoxy group at C-6 was replaced by an oxo group in four steps to give cycloketone 115 that was used for further construction of cyclic part of brassinosteroids (Scheme 13).
 |
| | Scheme 13 Synthesis of cycloketone 115 from stigmasterol 24. | |
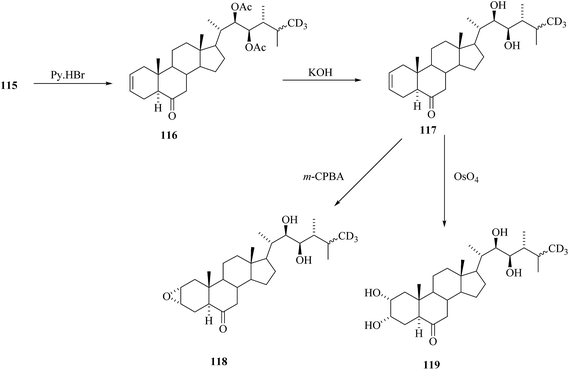
Treatment of compound 115 in boiling dimethyl acetamide in the presence of Py. HBr directed to the preparation of compound 116 containing 2,3 double bond in the ring A (Scheme 14).
 |
| | Scheme 14 Synthesis of 24-[26-2H3] epiCS 119. | |
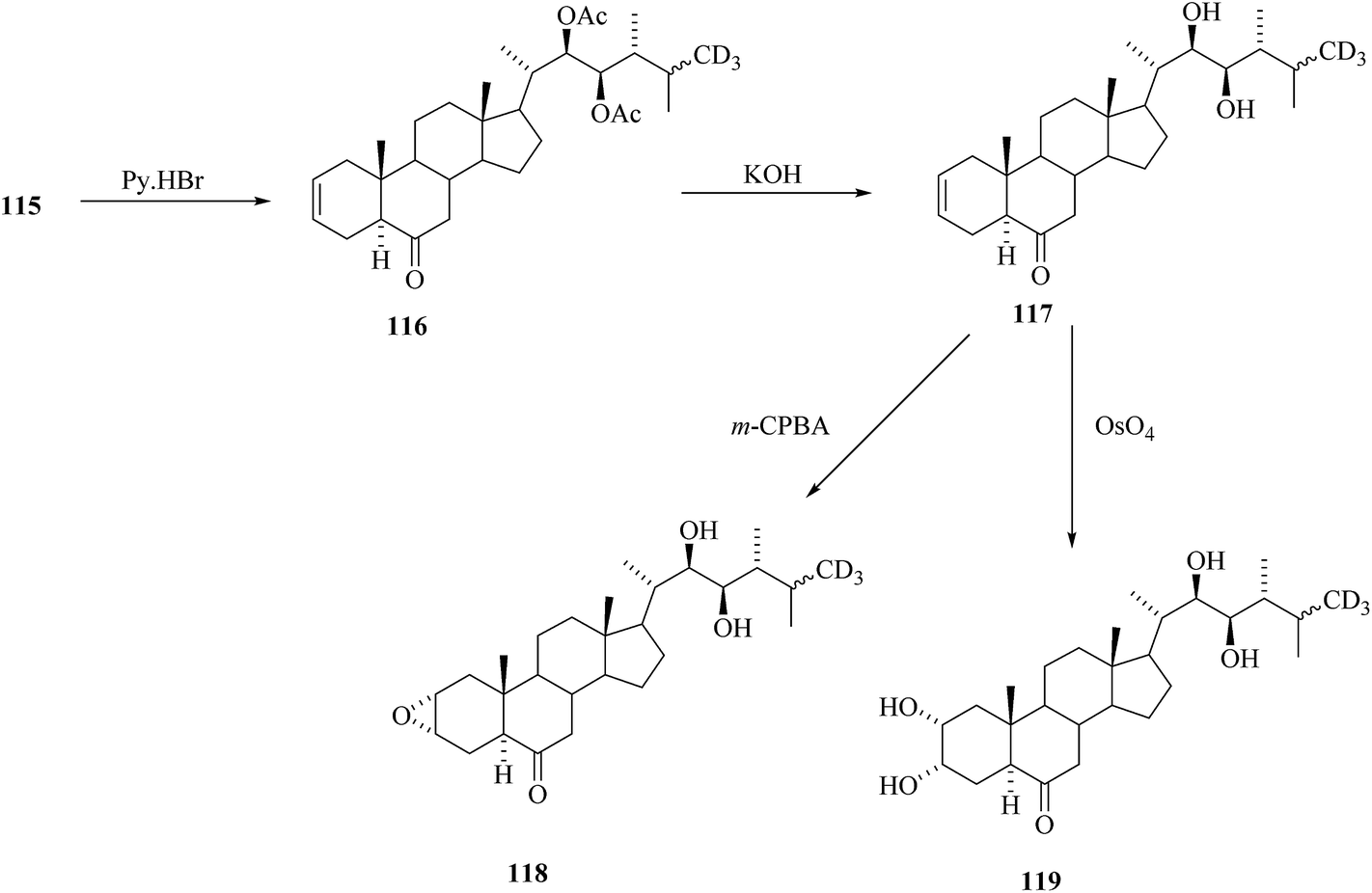
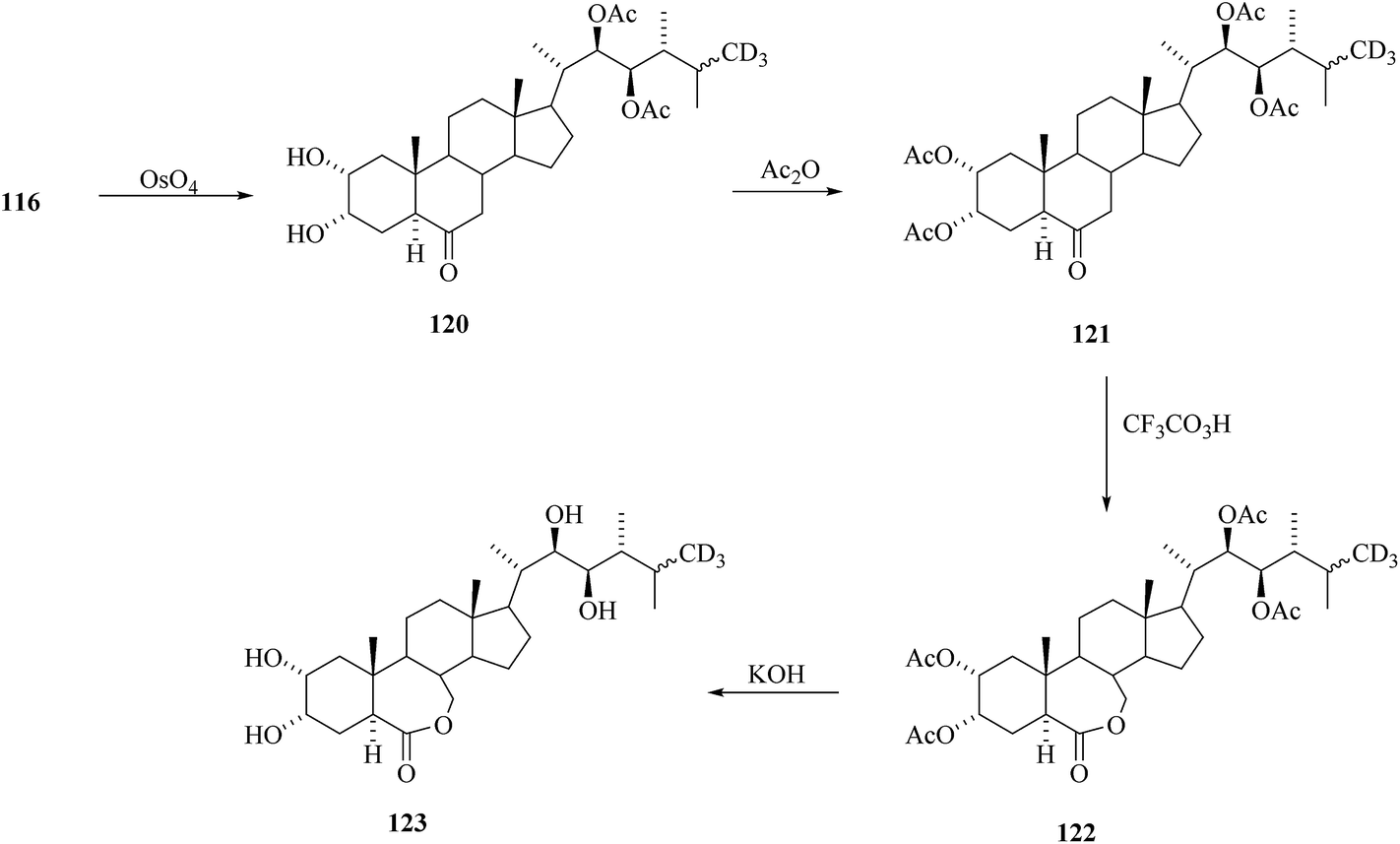
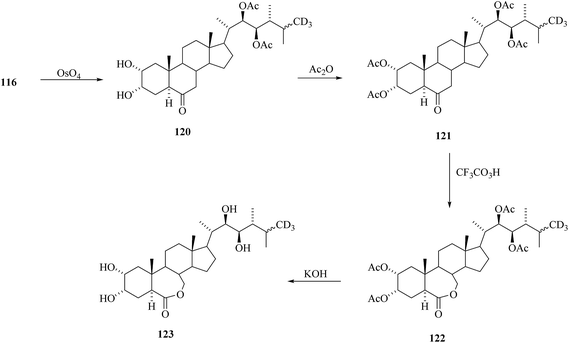
Removal of acetyl masking groups revealed the presence of 24-[26-2H3]episecasterol 117. Recently, 24-episecasterol was found cytotoxic against MCF-7 cells.44 Epoxidation of the olefin 117 gave deuterated epoxide 118 and dihydroxylation of olefin 117 gave 24-[26-2H3]epiCS 119 (Scheme 14). 24-[26-2H3]epiBL 123 was eventually prepared from protected 24-[26-2H3]episecasterol 116 (Scheme 15). The diacetate of [26-2H3] epics 120 obtained by dihydroxylation of 116 was acetylated and converted by Baeyer–Villiger oxidation to tetracetate of 24-[26-2H3]epiBL 121. After alkaline hydrolysis of acetate groups and acidic workup to restore the lactone ring in 24-[26-2H3] epibrassinolide 123. It is worth mentioning here that this paper is reporting the protocols enabling the direct Baeyer–Villiger oxidation of 24-epicastasterone to 24-epibrassinolide with more than satisfactory yields and without need of the protection of hydroxy groups.45,46
 |
| | Scheme 15 Synthesis of deuterated 24-[26-2H3] epiBL 123. | |
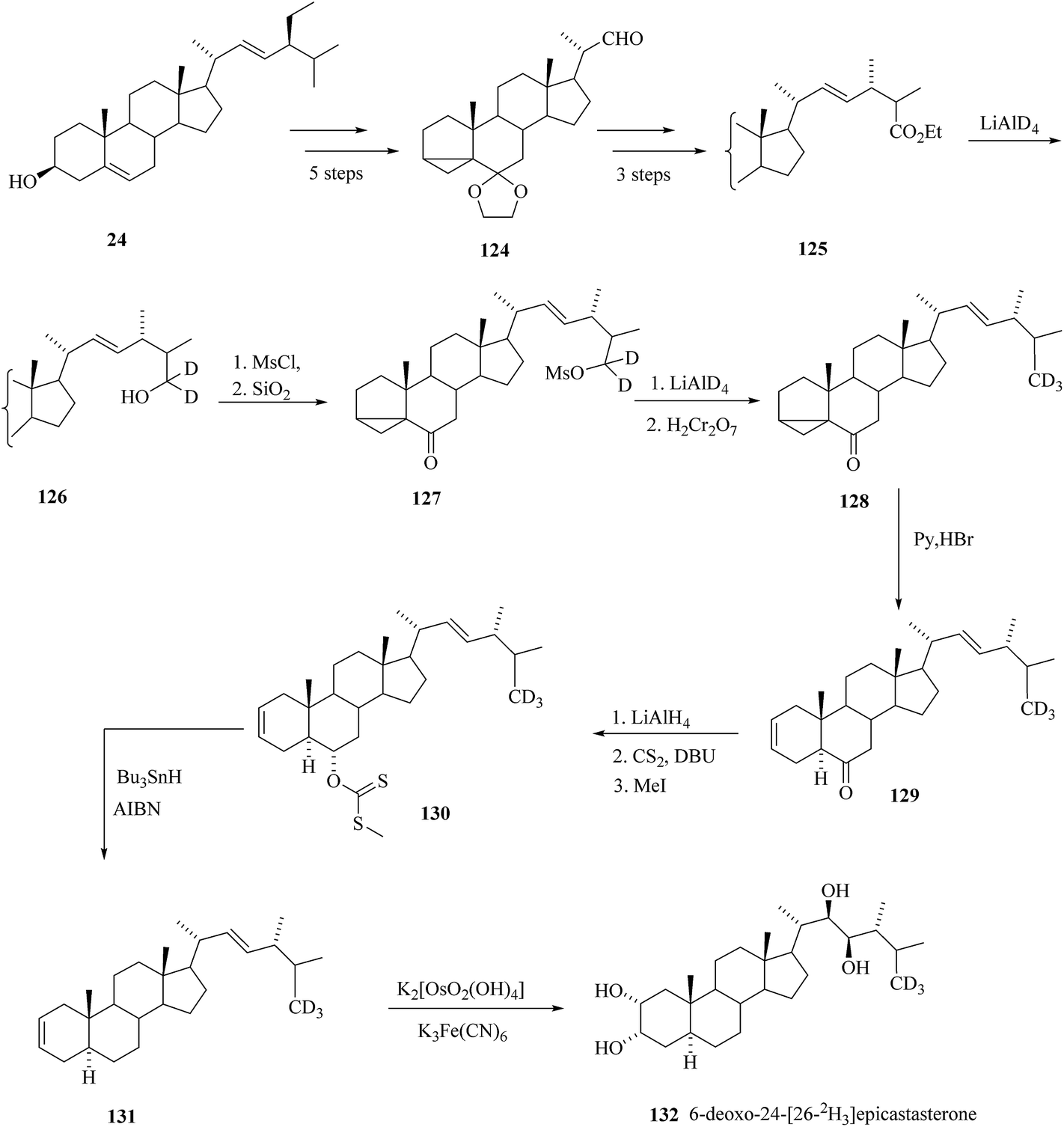
2.1.6. Synthesis of 6-deoxo-24-[26-2H3]epiCS.
The position and reaction sequence for introducing the label was selected based on the previous experience with the synthesis of deuterated BRs (Scheme 16).
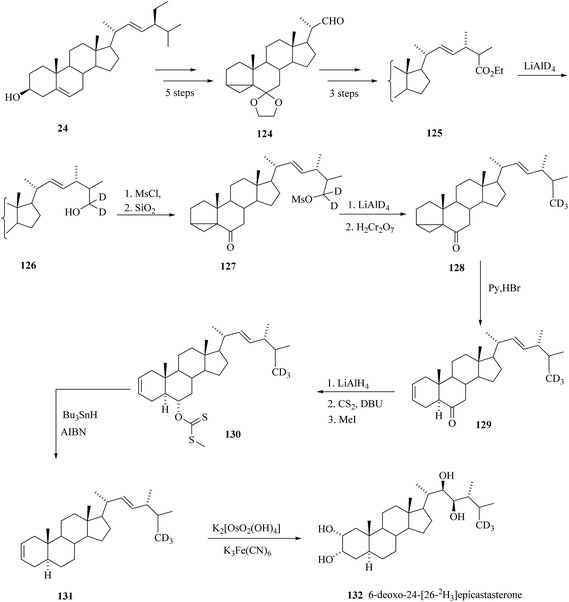 |
| | Scheme 16 Synthesis of 6-deoxo-24-[26-2H3] epiCS 132. | |
Stigmasterol 24 was a starting compound that was converted in five steps to 22-aldehyde 124. Claisen rearrangement was the key step in conversion of 22-aldehyde 124 to the ester 125. Using the reduction with LiAlD4-mesylation-reduction with LiAlD4 sequence as described above the ester group of 125 was converted to [2H3] methyl group in 128. Rearrangement of 3,5-cyclo derivative 128 to Δ2 derivative 129 was achieved by boiling with pyridinium hydrobromide. Deoxygenation of at C-6 was done via methyl xanthate 130 and for final cis hydroxylation potassium osmate was used. Thus, the 6-deoxo-24-[26-2H3]epiCS 132 was prepared47 in overall yield 18% calculated on ester 125.
2.2. BRs with deuterium label in ring A
2.2.1. Stereospecific labelling of brassinosteroids with hydrogen isotope.
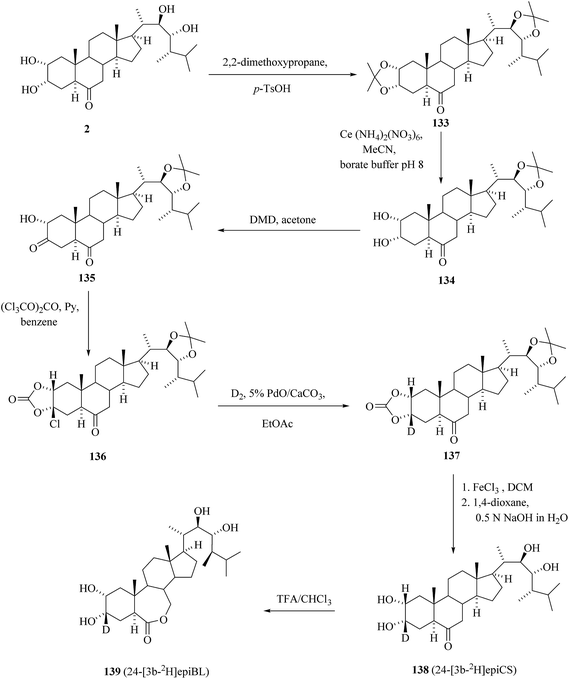
As the deuterium labelled BRs must have at least 4 deuterium atoms in metabolically stable positions to be useful as MS internal standards the deuterium labelling in ring A is not very attractive. On the other hand ring A is good target for labelling with tritum especially if the aim is to prepare suitable precursor for tritiation from the non-labelled target BR. It is the rule to model the tritiation reactions first with deuterium and therefore we developed recently general stereospecific method of introduction of deuterium to ring A of BRs having 2α,3α-dihydroxy group.
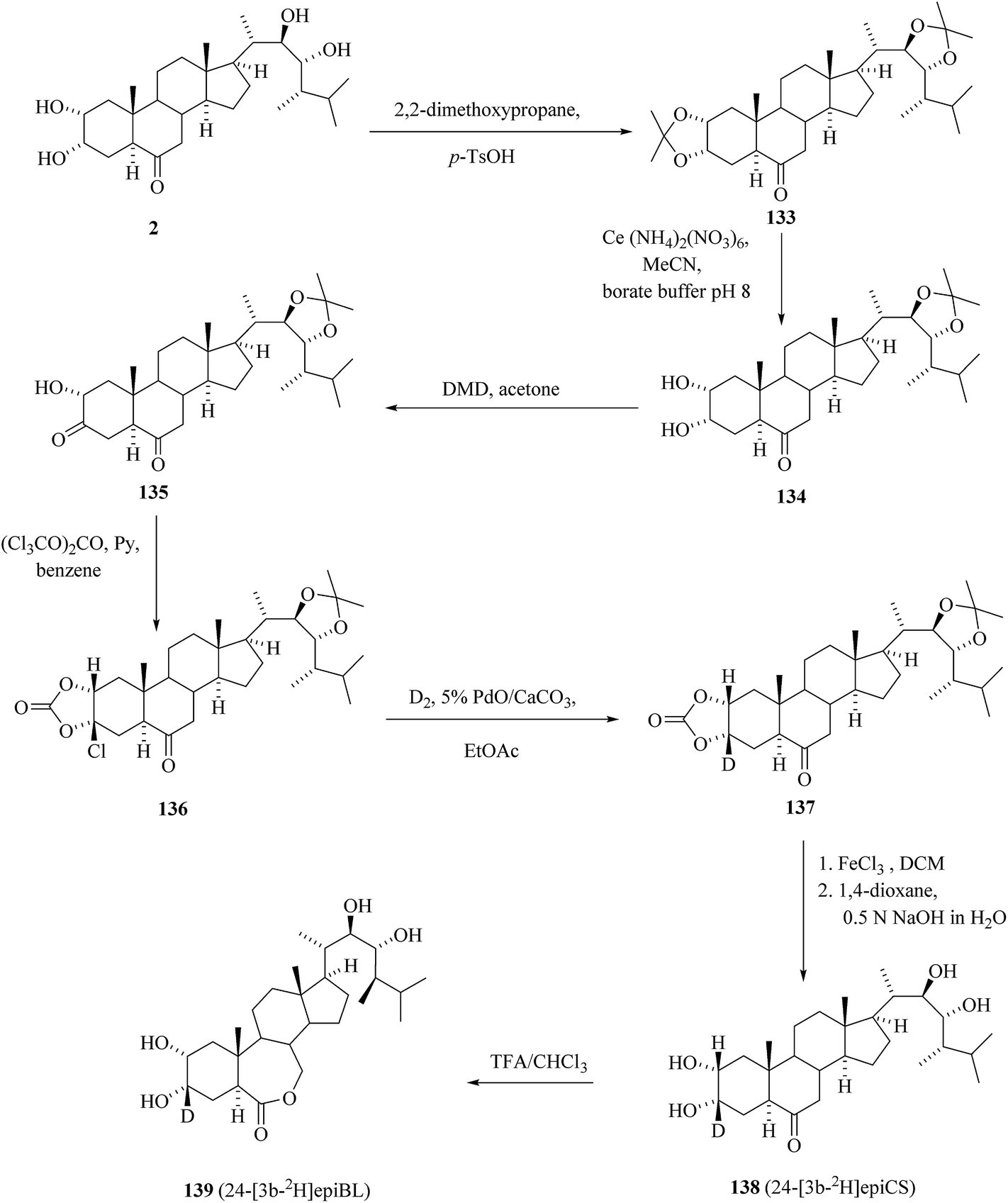
In quest for the suitable precursor for tritiation we discovered a stereospecific reaction of α-hydroxy ketone 135, prepared in three steps from 24-epicastasterone 2 in 47% yield, with triphosgene48 giving in 99% yield 3β-chloro-2α,3α-(carbonyldioxy) derivative 136 (Scheme 17).45 While catalytic reductive dechlorination is frequently used for the introduction of hydrogen isotopes on aromatic rings49 to our best knowledge there are no literature data available for either aliphatic or alicyclic substrates. Catalytic dehalogenation of chlorocarbonate 136 with deuterium gas on 5% PdO/CaCO3 in the presence of triethylamine in ethyl acetate afforded the 2α,3α-(carbonyldioxy)[3β-2H]derivative 137 in 65% yield and 80% isotopic enrichment. Removal of protecting groups in two-step one pot sequence gave 24-[3β-2H] epicastasterone 138 in 91% yield on 137. Baeyer–Villiger oxidation of [3β-2H]epiCs 138 with trifluoroperoxyacetic acid gave 24-[3β-2H]epiBL 139 in a 65% yield with no loss of deuterium label.
 |
| | Scheme 17 Synthesis of 24-[3β-2H]epiCS and 24-[3β-2H]epiBL. | |
2.3. BRs with deuterium label in ring B
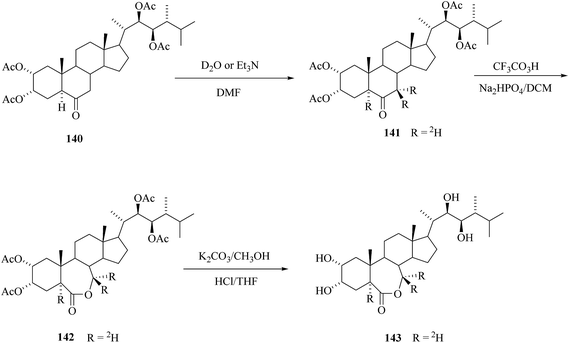
2.3.1. Synthesis of 24-[5,7,7-2H3]epiBL.
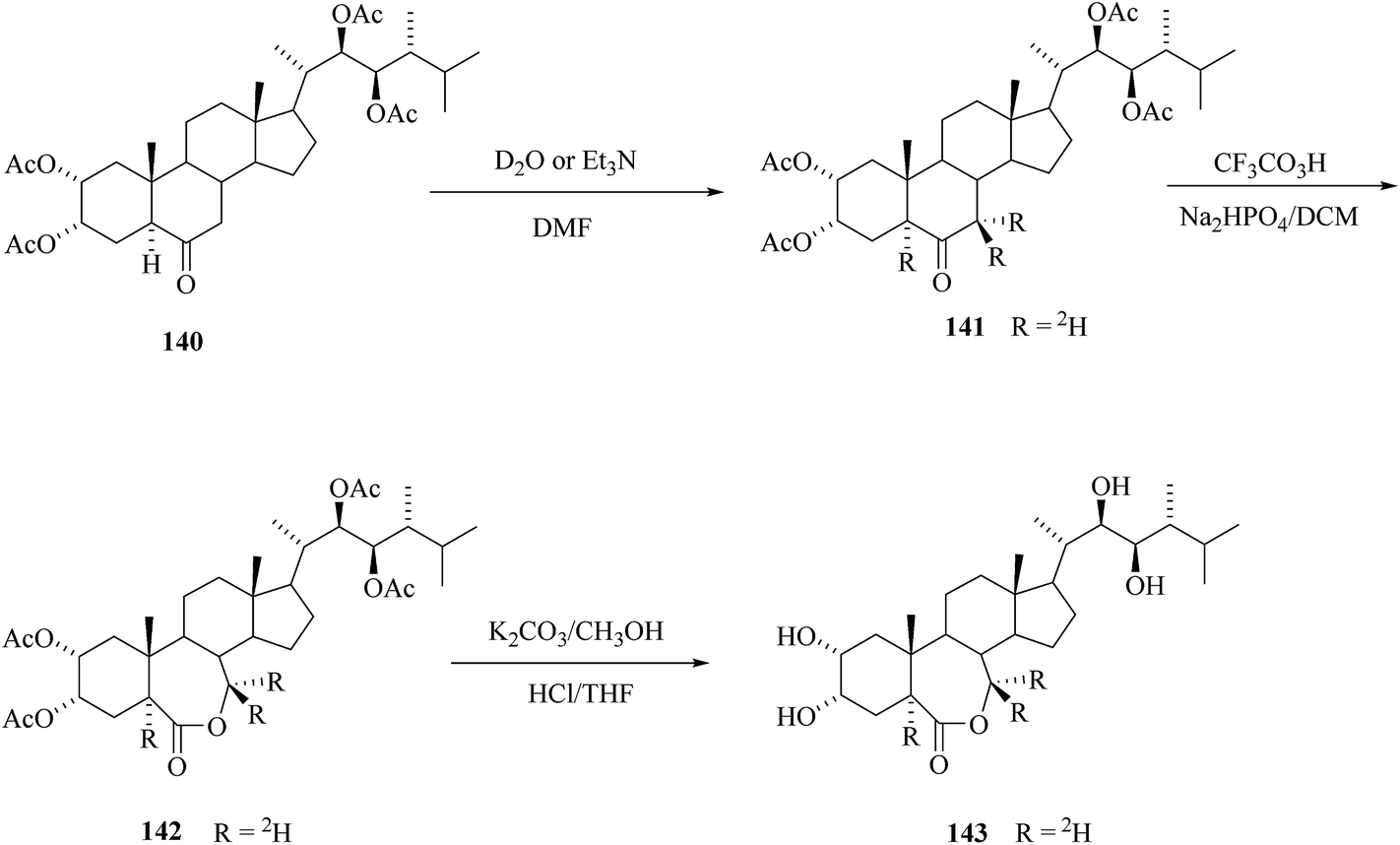
For BRs containing keto group the base catalyzed exchange of α-hydrogens with 2H2O can be exploited for deuterium introduction. Kolbe et al.20 reported the preparation of 24-[5,7,7-2H3]epiBL starting from tetracetate of 24-epiCS 140 (Scheme 18) or alternatively from 2,3,22,23-bis-isopropylidenedioxy-24-epiCS 133 (see Scheme 17). The exchange with 2H2O in DMF was catalyzed by triethyl amine. Bayer–Villiger oxidation with CF3CO3H of the obtained labelled 24-epiCS derivative 141 and its final deprotection gave 24-[5,7,7-2H3]epiBL 143. This procedure has the advantage of specific introduction of labelling at a late stage, whereas three deuterium atoms were introduced in a stable position at C-5 and C-7. A procedure reported by Allevi et al.50 is less convenient because it uses high excess of labelled water and methanol and there is the need of separation of side chain epimers.
 |
| | Scheme 18 Synthesis of 24-[5,7,7-2H3] epiBL. | |
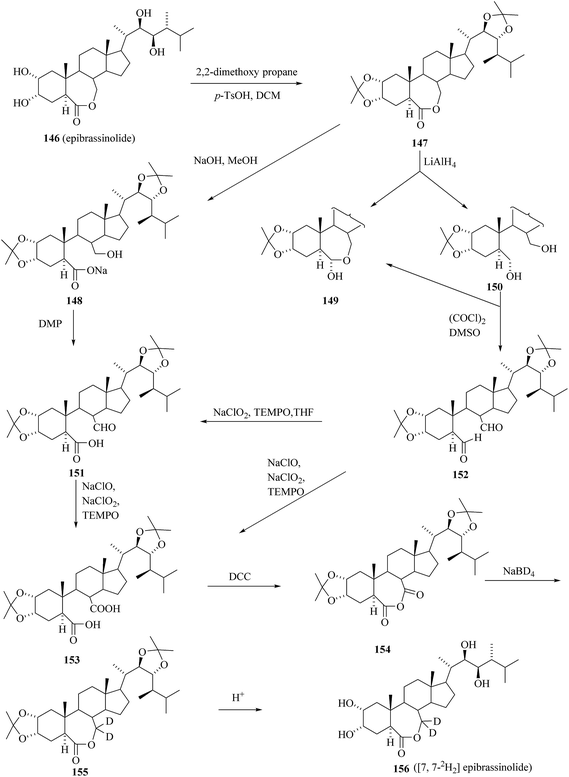
2.3.2. Synthesis of 24-[7,7-2H2]epiBL.
The intended transformation of lactone group in epibrassinolide 146 was carried out after protecting both diol functions by isopropylidenation. The key transformation is the preparation of 6,7-seco diacid 153 from diacetonide 147 in three step and then formation of the cyclic anhydride 154 from 6,7-seco diacid 153 (Scheme 19). Regioselective reduction of anhydride 154 with NaBD4 gave deuterated diacetonide 155. Acidic hydrolysis of acetonide groups on 155 provided [7,7-2H2]epiBL 156 with 82% isotopic enrichment.51
 |
| | Scheme 19 Synthesis of 24-[7,7-2H2]epiBL. | |
3. Synthesis of tritium-labelled BRs
3.1. Synthesis of 24-[5,7,7-3H3]epiBL
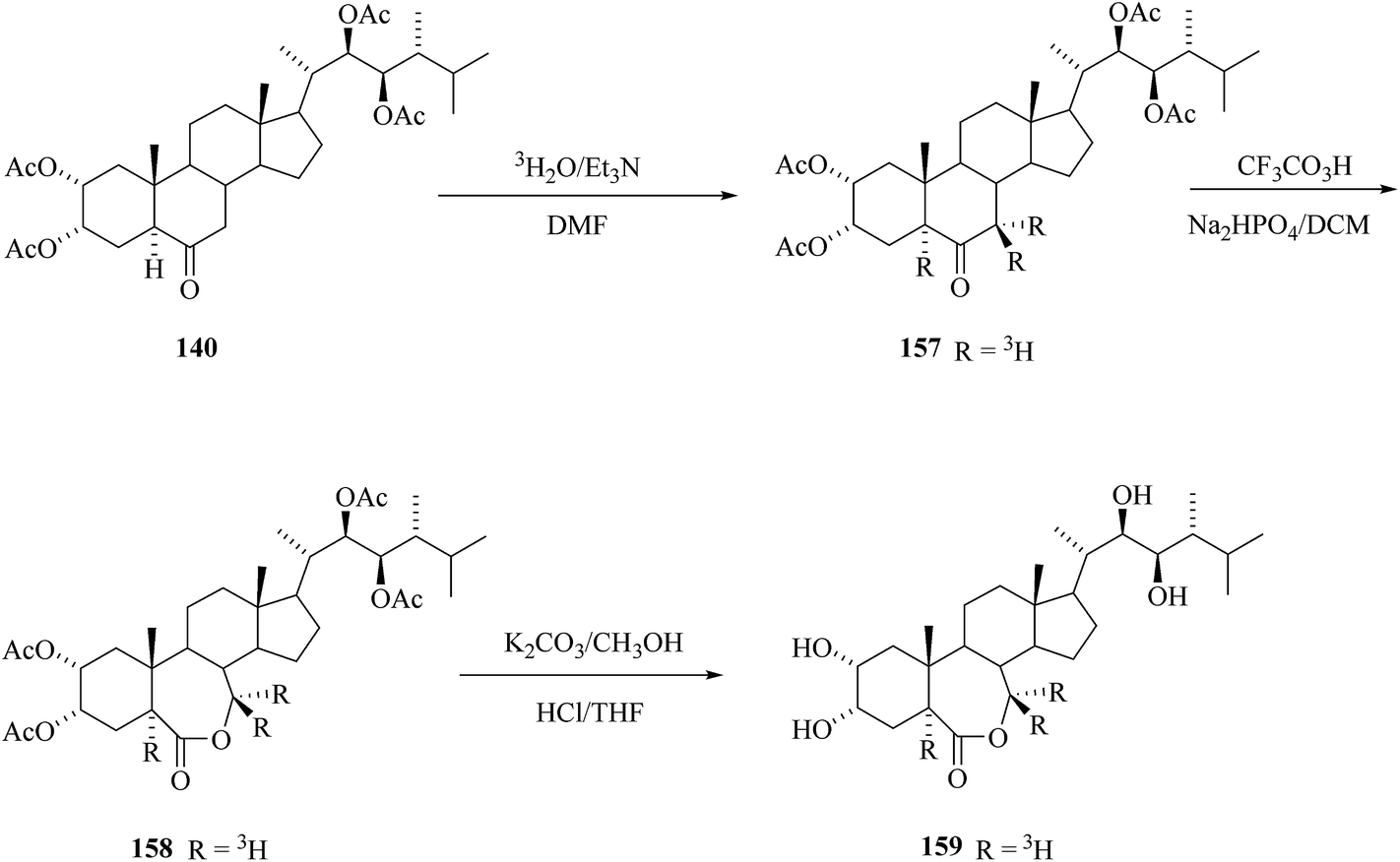
By the same method as described above for the preparation of corresponding deuterium labelled 24-epiBL 143 tritium was introduced to tetracetate of 24-epiCS 140 by base catalyzed exchange reaction with tritiated water having specific activity 1.1 Ci mL−1. After Bayer–Villiger oxidation of 157 and removal of acetyl protecting groups 24-[5,7,7-3H3] epiBL with specific activity (S.A.) 6 mCi mmol−1.20 The same sequence was performed also with 2,3,22,23-bis-isopropylidenedioxy-24-epiCS 133 as starting compound. The advantage of the use of isopropylidene protecting groups is the more simple deprotection step – the lactone ring is not opened under acidic hydrolysis conditions as the opposite is true for basic hydrolysis needed for the acetate group cleavage (Scheme 20).
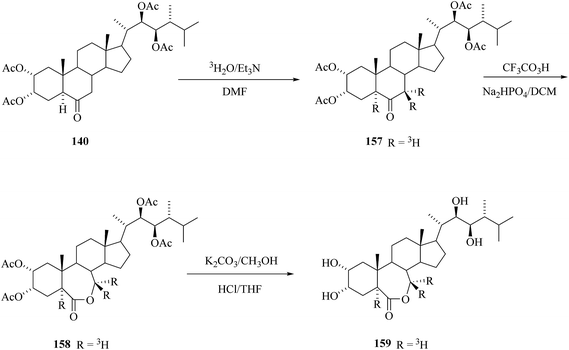 |
| | Scheme 20 Synthesis of 24-[5,7,7-3H3]epiBL. | |
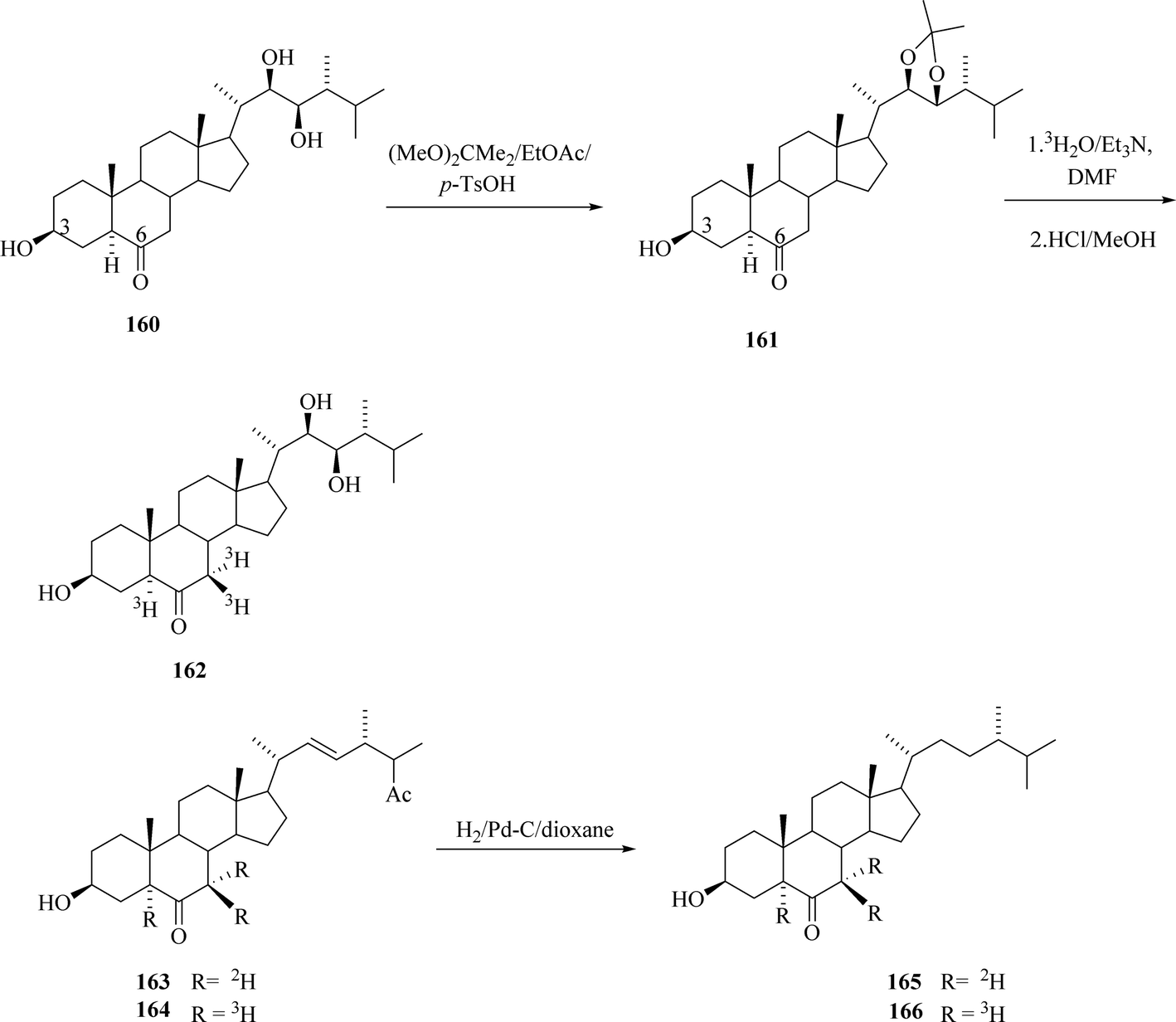
3.2. Synthesis of 24-[5,7,7-3H]epiTE, 6-oxo-24β-methyl-22-dehydro[5,7,7,3H]cholestanol and 6-oxo-24-[5,7,7-3H]epicampestanol
All title compounds were prepared by base catalysed exchange of α-keto hydrogens with tritiated water52 with S.A. = 0.8 Ci mmol−1 (Scheme 21). 24-[5,7,7-3H]epiteasterone 162 was prepared with S.A. = 1.5 mCi mmol−1 6-oxo-24β-methyl-22-dehydro[5,7,7,-3H] cholestanol 164 was labelled without protection of hydroxyl groups and its S.A. was not indicated. Cholestanol derivative 164 was further converted via hydrogenation of the double bond in the side chain catalysed by 10% Pd/C and then 6-oxo-24-[5,7,7-3H]epicampestanol purified by column chromatography had S.A. = 3.5 mCi mmol−1.
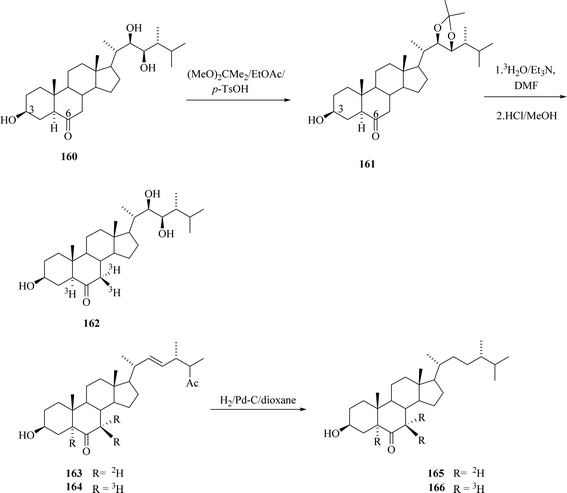 |
| | Scheme 21 Labelling of biogenetic BRs precursors. | |
4. Synthesis of 14C-labelled BRs
4.1. Synthesis of (22R,23R)-and (22S,23S)-24-[4-14C] epiBL
There is only one report available in the literature on 14C-labelled brassinosteroids so far. Seo et al.53 have reported the synthesis of [4-14C]-labelled epiBL 183 and 184. The C-4 position in epiBL 183 was selected for 14C labelling because of its stability to metabolic loss and easy way to do the preparation. According to the established method reported in the literature for incorporation of 14C into the C-4 position of steroids, the enol lactone 172 was synthesized from the starting material brassicasterol 167 in five steps (Scheme 22). This lactone 172 was then treated with [14C] methyl iodide (prepared by known method from barium [14C]carbonate via [14C]methanol) to give bridged ketone 173. Alkaline treatment of 173 in MeOH gave [4-14C]brassicasterone 174. Acetylation of 174 with isopropenyl acetate under acid catalysis gave the enol acetate 175 that was reduced with sodium borohydride in methanol to give [4-14C]brassicastero 176 as the major product 3,5-cyclo-6-ol 178 was obtained in 91.7% yield by mesylation to give 177 followed by treatment with sodium carbonate in acetone (Scheme 23). Jones oxidation of 178 gave 3,5-cyclo-6-one l79, which was treated with lithium bromide and camphor sulfonic acid in dimethyl acetamide to rearrange to 2,22-diene-6-one 180 in quantitative yield. Oxidation of 180 with osmium tetroxide gave a stereoisomeric mixture of 2,3,22,23-tetraols 181 and 182 which is separated through the repeated chromatography and recrsytallization. Bayer–Villiger oxidation with TFA in dichloromethane gave the (22R,23R)-7-oxa-lactone 183 contaminated with a small amount of its 6-oxa isomer. (22S,23S)-Tetraol also gave the (22S,23S)-7-oxa-lactone 184 contaminated with its 6-oxa isomer. The final products (22R,23R)-24-[4-14C]epiBL 183 and (22S,23S)-24-[4-14C]epiBL 183 were obtained in 3.20% and 4.46% radiochemical yield (based on Ba14CO3), respectively. Specific radioactivity of 183 and 183 was 56.8 mCi mmol−1.
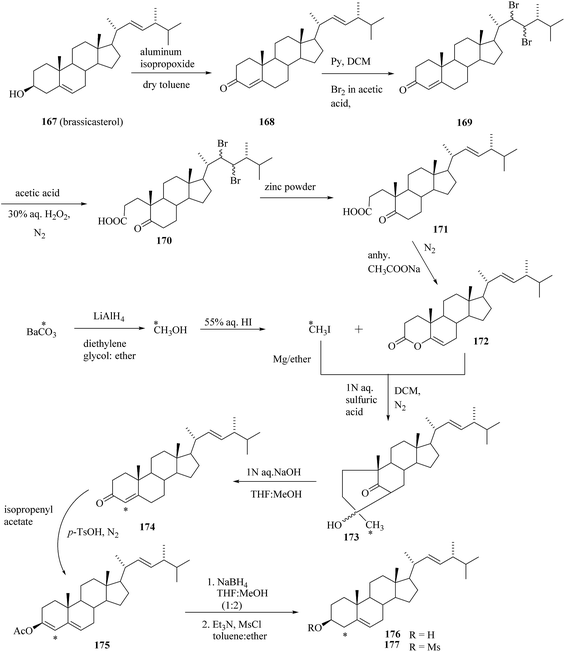 |
| | Scheme 22 Synthetic pathway to (22R,23R)-and (22S,23S)-24-[4-14C] epiBL I. | |
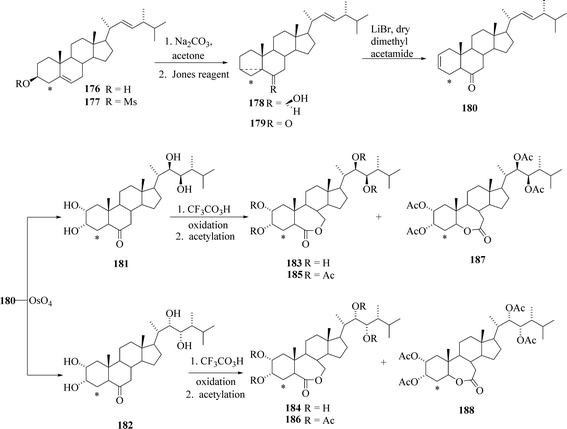 |
| | Scheme 23 Synthetic pathway to (22R,23R)-and (22S,23S)-24-[4-14C] epiBL II. | |
5. Summary
The number of published synthesis of BRs labelled either by stable isotopes or by radioisotopes reflects their importance for the biochemical studies of this interesting group of plant growth regulators. The main effort was devoted to the synthesis of multideuterated BRs in side chain as internal standards for MS. For labelling with tritium the methods developed for multideuterated BRs are not useful. The methods using exchange with tritiated water were described. However, of in this way prepared BRs have specific activities only in order of several mCi mmol−1. What remains to be done is to prepare the BRs ligands with the specific activities of order tens of Ci mmol−1 to enable the search for BRs receptors. Only one example of the synthesis of 14C-labelled BR reflects the shortage and fast increase of price of Ba14CO3 in recent years. Notwithstanding, some BRs are considered as potential drugs and if they will pass the preclinical sieve, there will be certainly need for their labelling with 14C.
List of abbreviations
| Ac2O | Acetic anhydride |
| AIBN | Azobisisobutyronitrile |
| Aq | Aqueous |
| BL | Brassinolide |
| BR | Brassinosteroid |
| BuLi | Butyllithium |
| CS | Castasterone |
| D | Deuterium |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DCC | Dicyclohexylcarbodiimide |
| DCM | Dichloromethane |
| (DHQ)2-PHAL | Dihydroquinine 1,4-phthalazinediyl diether |
| (DHQD)2-PHAL | Dihydroquinidine 1,4-phthalazinediyl diether |
| DMD | Dimethyldioxirane |
| DMF | Dimethylformamide |
| DMP | 4-Dimethylamino Pyridine |
| DMSO | Dimethyl sulfoxide |
| epiBL | epibrassinolide |
| Et3N | Triethyl amine |
| EtOAc | Ethyl acetate |
| Hg(OAc)2 | Mercury(II) acetate |
| LiAlD4 | Lithium Aluminum Deuteride |
|
m-CPBA |
meta-Chloroperoxybenzoic acid |
| MCF | Michigan cancer foundation |
| MeOH | Methanol |
| MsCl | Methanesulfonyl chloride |
| Na/Hg | Sodium amalgam |
| NBS |
N-Bromosuccinic imide |
| PCC | Pyridinium chlorochromate |
|
p-TsOH |
p-Toluenesulfonic acid |
| Py | Pyridine |
| T | Tritium |
| TE | Teasterone |
| TEMPO | 2,2,6,6-Tetramethylpiperidinyl-1-oxyl |
| TFD | Trifluromethyldioxirane |
| THF | Tetrahydrofuran |
| TsCl | 4-Toluenesulfonyl Chloride |
| TY | Typhasterol |
Acknowledgements
This work was realized at IOCB, Prague, Czech Republic and supported by Academy of Sciences of the Czech Republic (RVO 61388963), by Grant IAA400550801 of the Grant Agency of the Academy of Sciences of the Czech Republic and by CNMS, Jain University, Bangalore. The authors wish to express thanks to all for financial support.
References
- S. Fujioka and A. Sakurai, Nat. Prod. Rep., 1997, 14, 1–10 RSC.
- A. Bajguz and A. Tretyn, Phytochemistry, 2003, 62, 1027–1046 CrossRef CAS.
- V. Castilla, J. Ramirez and C. E. Coto, Curr. Med. Chem., 2010, 17, 1858–1873 CrossRef CAS.
- S. D. Clouse, Plant J., 1996, 10, 1–8 CAS.
-
V. A. Khripach, V. N. Zhabinski and A. de Groot, Brassinosteroids: A New Class of Plant Hormones, Academic Press, San Diego, 1999 Search PubMed.
-
Brassinosteroids: Steroidal Plant Hormones, ed. A. Sakurai, T. Yokota and S. D. Clouse, Springer, Tokyo, 1999 Search PubMed.
-
V. Marquardt and G. Adam, in Chemistry of Plant Protection, ed. W. Ebing, Springer, Berlin, 1991, vol. 7, pp. 103–139 and the references sited therein Search PubMed.
- H. Suzuki, T. Inoue, S. Fujioka, T. Saito, S. Takatsuto, T. Yokota, N. Murofushi, T. Yanagisawa and A. Sakurai, Photochemistry, 1995, 40, 1391–1397 CrossRef CAS.
- S. Fujioka, T. Inoue, S. Takatsuto, T. Yanagisawa, T. Yokota and A. Sakurai, Biosci., Biotechnol., Biochem., 1995, 59, 1543–1547 CrossRef CAS.
- S. Takatsuto, C. Gotoh, T. Noguchi, T. Nomura, S. Fujioka and T. Yokota, J. Chem. Res., Synop., 1998, 206–207 RSC.
- H. Suzuki, S. Fujioka, S. Takatsuto, T. Yokota, N. Murofushi and A. Sakurai, J. Plant Growth Regul., 1994, 13, 21–26 CrossRef CAS.
- A. Sakurai and S. Fujioka, Biosci., Biotechnol., Biochem., 1997, 61, 757–762 CrossRef CAS PubMed.
- S. Fujioka, J. Li, Y.-H. Choi, H. Seto, S. Takatsuto, T. Noguchi, T. Watanabe, H. Kuriyama, T. Yokota, J. Chory and A. Sakurai, Plant Cell, 1997, 9, 1951–1962 CrossRef CAS PubMed.
-
T. Noguchi, S. Fujioka, S. Takatsuto, S. Yoshida, A. Sakurai, J. Li and J. Chory, Abstract Papers of 32nd Annual Meeting of The Japanese Society for Chemical Regulation of Plants, Tokyo, 1997, p. 93.
- S. Fujioka and A. Sakurai, Physiol. Plant., 1997, 100, 710–715 CrossRef CAS PubMed.
- T. Yokota, Trends Plant Sci., 1997, 2, 137–143 CrossRef.
- A. Antonchick, A. Svatoš, B. Schneider, O. V. Konstantinova, V. N. Zhabinskii and V. A. Khripach, Phytochemistry, 2005, 66, 65–72 CrossRef CAS PubMed.
- S. Takatsuto and N. Ikekawa, Chem. Pharm. Bull., 1984, 32, 2001–2004 CrossRef CAS.
-
(a) J. Malíková, J. Swaczynová, Z. Kolář and M. Strnad, Phytochemistry, 2008, 69, 418–426 CrossRef PubMed;
(b) J. Steigerová, J. Oklešťková, M. Levková, L. Rárová, Z. Kovář and M. Strnad, Chem.-Biol. Interact., 2010, 188, 487–496 CrossRef PubMed.
- A. Kolbe, V. Marquardt and G. Adam, J. Labelled Compd. Radiopharm., 1992, 31, 801–805 CrossRef CAS PubMed.
- V. A. Khripach, V. N. Zhabinskii, O. V. Gulyakevich, O. V. Konstantinova, A. Y. Misharin, A. R. Mekhtiev, V. P. Timofeev and Y. V. Tkachev, Russ. J. Bioorg. Chem., 2010, 36, 746–754 CrossRef CAS.
- S. Takatsuto and N. Ikekawa, Chem. Pharm. Bull., 1986, 34, 4045–4049 CrossRef CAS.
- S. C. Lee, S. Joo, C. Park, S. Son, J. Youn, M. Kim, S. Jeong and S. Kim, Bull. Korean Chem. Soc., 2011, 32, 332–334 CrossRef CAS.
-
G. Adam, A. Porzel, J. Schmidt, B. Schneider and B. Voigt, in Studies in Natural Products Chemistry, ed. Atta-ur-Rahrnan, Elsevier, Amsterdam, 1996, vol. 18, pp. 495–549 Search PubMed.
- M. D. Grove, G. F. Spencer, W. K. Rohwedder, N. Mandava, J. F. Worley, J. D. Warthen, G. L. Steffens, J. L. Flippen-Anderson and J. C. Cook, Nature, 1979, 225, 1065–1066 Search PubMed.
- S. Takatsuto and N. Ikekawa, Chem. Pharm. Bull., 1986, 34, 1415–1418 CrossRef.
- S. Takatsuto and N. Ikekawa, Chem. Pharm. Bull., 1986, 34, 4045–4049 CrossRef CAS.
- S. Takatsuto and N. Ikekawa, J. Chem. Soc., Perkin Trans. 1, 1986, 591–593 RSC.
- V. A. Khripach, V. N. Zhabinskii, O. V. Konstantinova and N. B. Khripach, Tetrahedron Lett., 2000, 41, 5765–5767 CrossRef CAS.
- V. A. Khripach, V. N. Zhabinskii, O. V. Konstantinova, N. B. Khripach, A. P. Antonchick and B. Schneider, Steroids, 2002, 67, 587–595 CrossRef CAS.
- K. Mori, M. Sakakibara, Y. Ichikawa, H. Ueda, K. Okada, T. Umemura, G. Yabuta, S. Kuwahara, M. Kondo, M. Minobe and A. Sogabe, Tetrahedron, 1982, 38, 2099–2109 CrossRef CAS.
- V. A. Khripach, V. N. Zhabinskii, V. K. Olkhovick and F. A. Lakhvich, Zh. Org. Khim., 1990, 26, 1966–1976 CAS.
- J. A. Steele and E. Mosettig, J. Org. Chem., 1963, 28, 571–572 CrossRef CAS.
- J. J. Pappas, W. P. Keaveney, E. Gancher and M. Berger, Tetrahedron Lett., 1966, 7, 4273–4278 CrossRef.
- A. P. Antonchick, B. Schneider, V. N. Zhabinskii and V. A. Khripach, Steroids, 2004, 69, 617–628 CrossRef CAS PubMed.
- V. A. Khripach, V. N. Zhabinskii, A. P. Antonchick, O. V. Konstantinova and B. Schneider, Steroids, 2002, 67, 1101–1108 CrossRef CAS.
- M. Anastasia, P. Allevi, P. Ciuffreda and A. Fiecchi, J. Chem. Soc., Perkin Trans. 1, 1983, 2365–2367 RSC.
- M. Anastasia, P. Ciuffreda, M. Delpuppo and A. Fiecchi, J. Chem. Soc., Perkin Trans. 1, 1983, 379–382 RSC.
- M. Anastasia, P. Allevi, P. Ciuffreda, A. Fiecchi, P. Gariboldi and A. Scala, J. Chem. Soc., Perkin Trans. 1, 1985, 595–599 RSC.
- M. Koizumi, M. Ishiguro, M. Yasuda and N. Ikekawa, J. Chem. Soc., Perkin Trans. 1, 1983, 1401–1410 RSC.
-
V. Marquardt and G. Adam, Chemistry of plant protection, ed. W. Ebing, Springer, Berlin, 1991, vol. 7, p. 103 Search PubMed.
- H. Seto, S. Fujioka, H. Koshino, S. Yoshida, T. Watanabe and S. Takatsuto, Tetrahedron Lett., 1998, 39, 7525–7528 CrossRef CAS.
- A. Kolbe, A. Porzel, J. Schmidt and G. Adam, J. Labelled Compd. Radiopharm., 2003, 46, 231–242 CrossRef CAS PubMed.
- V. A. Khripach, V. N. Zhabinskii, O. V. Gulyakevich, O. V. Konstantinova, A. Y. Misharin, A. R. Mekhtiev, V. P. Timofeev and Y. V. Tkachev, Russ. J. Bioorg. Chem., 2010, 36, 746–754 CrossRef CAS.
- A. Marek, M. R. Patil, B. Klepetářová, L. Kohout and T. Elbert, Tetrahedron Lett., 2012, 53, 2048–2050 CrossRef CAS PubMed.
- L. Kohout, Collect. Czech. Chem. Commun., 1994, 59, 457–460 CrossRef CAS.
- V. A. Khripach, V. N. Zhabinskii and O. V. Gulyakevich, Chem. Nat. Compd., 2012, 48, 601–605 CrossRef CAS.
- H. Eckert and B. Forster, Angew. Chem., Int. Ed. Engl., 1987, 26, 894–895 CrossRef PubMed.
-
R. Voges, J. R. Heys and T. Moenius, Preparation of compounds labelled with tritium and Carbon-14, John Wiley & Sons, Chichester, 2009, pp. 133–144 Search PubMed.
- P. Allevi, M. Anastasia, R. Cerana, P. Ciuffreda and P. Lado, Phytochemistry, 1988, 27, 1309–1313 CrossRef CAS.
- V. A. Khripach, N. B. Khripach, V. N. Zhabinskii, Y. Y. Zhiburtovich, B. Schneider and A. De Groot, J. Labelled Compd. Radiopharm., 2007, 50, 1153–1158 CrossRef CAS PubMed.
- A. Kolbe, B. Schneider, B. Voigt and G. Adam, J. Labelled Compd. Radiopharm., 1998, 41, 131–137 CrossRef CAS.
- S. Seo, T. Nagasaki, Y. Katsuyama, F. Matsubara, T. Sakata, M. Yoshioka and Y. Makisumi, J. Labelled Compd. Radiopharm., 1989, 27, 1383–1393 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a