
High loading of doxorubicin into styrene-terminated porous silicon nanoparticles via π-stacking for cancer treatments in vitro†
Abstract
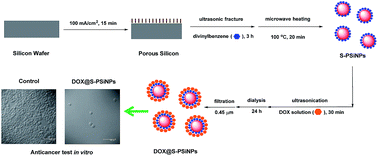
Porous silicon nanoparticles (PSiNPs) as nanocarriers for anticancer drug delivery have an important potential for cancer treatments, thus the development of PSiNPs-based delivery systems with efficient loading and controlled release of therapeutic drugs is necessary. Here, we present a novel strategy of incorporating doxorubicin (DOX) into styrene-terminated PSiNPs (S-PSiNPs) via π-stacking to form DOX@S-PSiNPs nanocomposites, which have a high-loading amount of DOX molecules. In addition, pH-controlled release of DOX molecules from the as-prepared DOX@S-PSiNPs nanocomposites was also observed. After cellular internalization of DOX@S-PSiNPs, the DOX molecules could be also efficiently released in the cytoplasm of cancer cells and then translocated into cellular nuclei, which prolonged their anticancer performance, compared with free DOX molecules.
 Please wait while we load your content...
Please wait while we load your content...

