
Cr3+-substitution induced structural reconfigurations in the nanocrystalline spinel compound ZnFe2O4 as revealed from X-ray diffraction, positron annihilation and Mössbauer spectroscopic studies†
Abstract
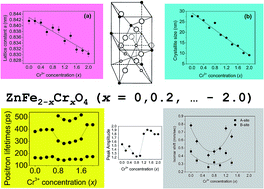
In an earlier work, the substitution of Zn2+ ions at the tetrahedral sites of nanocrystalline zinc ferrite (ZnFe2O4) by Ni2+ ions had been observed to cause a transformation from the normal spinel structure to the inverse one. The present study has been undertaken to explore the possibility of a similar change when the Fe3+ ions at the octahedral sites are replaced by Cr3+ ions. Concomitant lattice contraction and a steady decrease of the sizes of the nanocrystallites preceded and then resulted into the inversion of ZnFe2−xCrxO4 from normal spinel to inverse at x ≥ 0.8. Positron lifetime and coincidence Doppler broadening spectroscopic studies were carried out on the samples and a distinct third positron lifetime component emerged in the range of Cr3+ concentration 0.8 ≤ x ≤ 1.6. The new positron trapping sites were the result of the inversion of the spinel structure wherein the Cr3+ ions which substituted the Fe3+ ions at the octahedral sites got shifted to the tetrahedral sites, interchanging their positions with the Zn2+ ions. The incomplete success of inversion led to the generation of vacancy-type defects, which significantly trapped the positrons and the changes in their lifetimes indicated the occurrence of the process. The continued lattice contraction ensured an inverted spinel structure even for the final ZnCr2O4, which in coarse-grained form and at room temperature is a normal spinel. Mőssbauer spectroscopic studies also supported the idea of spinel inversion above x = 0.8 through definite changes in the isomer and quadrupole shifts.
 Please wait while we load your content...
Please wait while we load your content...

