
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral alkaline earth metal complexes with M–Se direct bond (M = Mg, Ca, Sr, Ba): syntheses, structures and ε-caprolactone polymerisation†
Ravi K. Kottalanka,
Adimulam Harinath and
Tarun K. Panda*
Department of Chemistry, Indian Institute of Technology Hyderabad, Ordnance Factory Estate, Yeddumailaram 502205, Telangana, India. E-mail: tpanda@iith.ac.in; Fax: +91 40 2301 6032; Fax: +91 40 2301 6036
First published on 20th April 2015
Abstract
We report here a series of enantiomeric pure alkaline earth metal complexes, each with a metallic direct bond of selenium, with {HN(R-*CHMePh)(P(Se)Ph2)} (1a) and {HN(S-*CHMePh)(P(Se)Ph2)} (1b), synthesised using two routes. The first route involves a trans metalation reaction of enantiomeric pure potassium phosphinoselenoic amide [K{N(R-*CHMePh)(Ph2P(Se))}{THF}n] (2a) or [K{N(S-*CHMePh)(Ph2P(Se))}{THF}n] (2b) prepared from the reaction between either 1a or 1b and [KN(SiMe3)2], and the corresponding alkaline earth metal diiodies in THF at room temperature to afford the enantiomeric pure complexes of composition [M{N(R-*CHMePh)P(Se)Ph2}2(THF)n] [M = Mg (3a), n = 1; M = Ca (4a), Sr (5a) and Ba (6a), n = 2] and [M{N(S-*CHMePh)P(Se)Ph2}2(THF)n] [M = Mg (3b), n = 1; M = Ca (4b), Sr (5b) and Ba (6b), n = 2]. The same heavier alkaline earth metal complexes (4a–6a and 4b–6b) can also be obtained through the silylamine elimination method using the corresponding metal bis(trimethylsilyl)amides [M{N(SiMe3)2}2(THF)n] (M = Ca, Sr, Ba) with phosphinoselenoic amine ligands 1a and 1b in ambient conditions. The solid-state structures of the metal complexes 4a–6a and 4b–6b were established using single-crystal X-ray diffraction analysis. In the solid state, all the metal complexes crystallise in the monoclinic P21 space group and each phosphinoselenoic amido ligand is ligated to the metal ion in a bidentate fashion. We also report the syntheses and structures of chiral amidophosphine-borane ligands {HN(R-*CHMePh)(P(BH3)Ph2)} (7a) and {HN(S-*CHMePh)(P(BH3)Ph2)} (7b) and the corresponding homoleptic barium complexes of composition [Ba{N(R-*CHMePh)P(BH3)Ph2}2(THF)2] (8a) and [Ba{N(R-*CHMePh)P(BH3)Ph2}2(THF)2] (8b). The molecular structures of 8a and 8b in the solid state confirm the attachment of chiral amidophosphine-borane ligands to the barium ions. The complexes 5 and 6 were tested as catalysts for the ring-opening polymerisation of ε-caprolactone. High activity in relation to the barium complexes 6a and 6b is observed, with moderate to narrow polydispersity index.
Introduction
Efficient synthesis of optically active compounds is one of the most important tasks of synthetic organic chemistry. The most promising methodology is catalytic asymmetric synthesis using a chiral metal centre. Among many useful metal species, alkaline earth metals have long been recognised as belonging to a class of less toxic and less harmful metals.1,2 However, besides the potential high utility of the alkaline earth species as a homogeneous catalyst for ring-opening polymerisation of various cyclic esters,3,4 polymerisation of styrene and dienes,5 and hydroamination and hydrophosphination reactions of alkenes and alkynes,6 its use in synthetic organic chemistry, especially in asymmetric synthesis as chiral catalyst, has been quite limited when compared to transition metal catalysts.1,2 Recently it was revealed that several catalytic asymmetric carbon–carbon bond-forming and related reactions proceeded smoothly in high enantioselectivites with the use of chiral Ca, Sr, and Ba catalysts.7–10 Their strong Brønsted basicity and mild Lewis acidity are promising and attractive characteristics and can influence their catalytic activity as well as their chiral modification capability in a positive manner.A wide variety of chiral phosphorus ligands have been prepared over the years, and their coordination chemistry with various metal ions has been studied extensively.11 In homogeneous catalyses, bidentate phosphine ligands, especially those having C2 symmetry, have usually been employed. In most cases the stereogenic centres are chiral phosphorus atoms or phosphines with chiral hydrocarbon substituents as derivatives of the chiral pool.11a The synthesis and limited use of heteroatom-substituted phosphines and their transition metal complexes have received some attention lately as a result of the search for new structural diversity.11b However little has been published on the use of chiral amines as backbone for chiral phosphorus ligands. These chiral P, N ligands, which usually coordinate via the phosphorus atom to the centre metal, were basically used in transition metal and rare earth metal chemistry.12
Recently we introduced various amidophosphine chalcogenide and borane ligands with P, N, E (E = O, S, Se, BH3) as donor atoms, into alkaline earth metal chemistry to study their coordination properties.13 These unique ligands are potentially capable of coordinating through the hard nitrogen and phosphorus donor atoms as well as the soft E donor atom. Bearing these characteristic features in mind, as well as our continuing interest in highly electropositive alkaline earth metals, catalytic activity and the vast potential of the field in asymmetric synthesis, we proposed to synthesise various novel chiral alkaline earth metal complexes stabilised by chiral amidophosphine selenoids and boranes, to explore the chemistry of alkaline earth metals in asymmetric synthesis. To achieve our target compounds with high-purity and good yield, we chose chiral phosphineamines HN(R-*CHMePh)(PPh2) and HN(S-*CHMePh)(PPh2), which were originally introduced by Brunner into coordination chemistry of the late transition metals.14 Roesky et al. introduced the same ligands into zirconium chemistry,15 group 3 and lanthanide chemistry.16 We synthesise the corresponding enantiomeric pure amidophosphine-selenoids [HN(R-*CHMePh)P(Se)Ph2] (1a) and [HN(S-*CHMePh)P(Se)Ph2] (1b) in order to introduce them into the alkaline earth metal chemistry. We envisage that these ligands potentially coordinate through the amido nitrogen and selenium atoms, thus forming a four-membered metallacycle with a centre metal ion.
In this context, detailed synthetic and coordination properties of homoleptic alkaline earth metal complexes of molecular composition [M{N(R-*CHMePh)-P(Se)Ph2}2(THF)2] [M = Mg (3a), Ca (4a), Sr (5a) and Ba (6a)] and [M{N(S-*CHMePh)P(Se)Ph2}2(THF)2] [M = Mg (3b), Ca (4b), Sr (5b) and Ba (6b)], were described with the chiral phosphinoselenoic amide ligands {HN(R-*CHMePh) (P(Se)Ph2)} (1a) and {HN(S-*CHMePh)(P(Se)Ph2)} (1b). In addition, we report the synthesis and structures of the chiral amidophosphine-borane ligands {HN(R-*CHMePh)(P(BH3)Ph2)} (7a) and {HN(S-*CHMePh)(P(BH3)Ph2)} (7b) and their corresponding homoleptic barium complexes of composition [Ba{N(R-*CHMePh)P(BH3)Ph2}2(THF)2] (8a) and [Ba{N(R-*CHMePh)P(BH3)Ph2}2(THF)2] (8b). The details of the ring-opening polymerisation of ε-caprolactone using complexes 5 and 6 are also presented.
Results and discussion
To introduce the chiral phosphinoselenoic amide ligands {HN(R-*CHMePh)(P(Se)Ph2)}− and {HN(S-*CHMePh)(P(Se)Ph2)}− into alkaline earth metal chemistry, we first synthesised the protio ligands {HN(R-*CHMePh)(P(Se)Ph2)} (1a) and {HN(S-*CHMePh)(P(Se)Ph2)} (1b) and their potassium salts [K{N(R-*CHMePh)(Ph2P(Se))}{THF}n] (2a) and [K{N(S-*CHMePh)(Ph2P(Se))}{THF}n] (2b). The potassium salts 2a and 2b were reacted with alkaline earth metal diiodide (MI2) to obtain corresponding homoleptic complexes.Chiral phosphinoselenoic amide ligands
The chiral phosphinoselenoic amides {HN(R-*CHMePh)(Ph2P(Se))} (1a) and {HN(S-*CHMePh)(Ph2P(Se))} (1b) were prepared in enantiomeric pure forms in a similar method as analogous [Ph2P(Se)NHCHPh2] and [Ph2P(Se)NHCPh3] were prepared, that is, they were synthesised in quantitative yield by the treatment of pure 1,1-diphenyl-N-(1-phenylethyl)phosphinamines {HN(R-*CHMePh)(Ph2P)} and {HN(S-*CHMePh)(Ph2P)} with slight excess elemental selenium in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2 molar ratio at ambient temperature in THF solvent (see Scheme 1).13c,17 Both the enantiomeric pure compounds 1a and 1b were characterised using standard 1H, 13C{1H}, 31P{1H} NMR spectra, combustion analysis and the solid-state structures were established using single-crystal X-ray diffraction analysis.
:1.2 molar ratio at ambient temperature in THF solvent (see Scheme 1).13c,17 Both the enantiomeric pure compounds 1a and 1b were characterised using standard 1H, 13C{1H}, 31P{1H} NMR spectra, combustion analysis and the solid-state structures were established using single-crystal X-ray diffraction analysis.
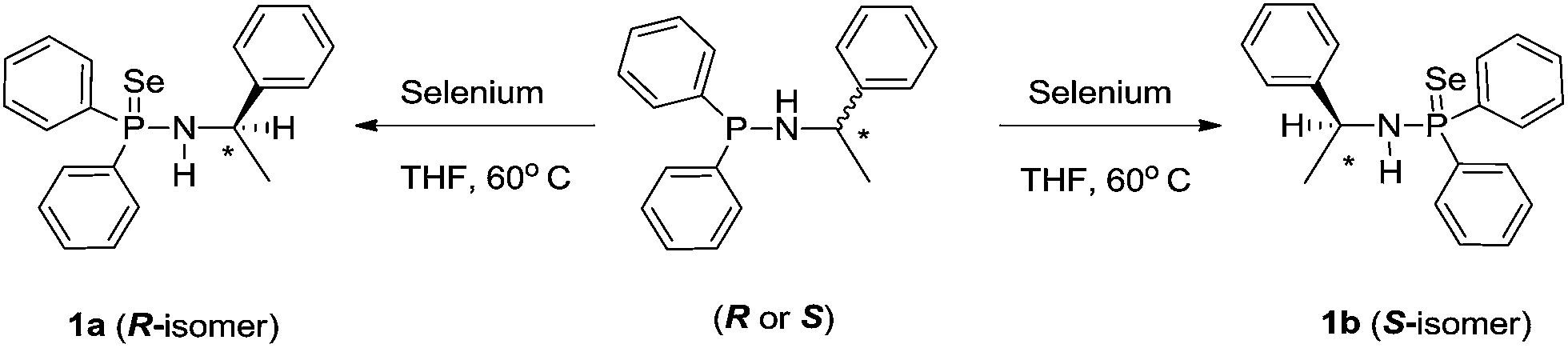 | ||
| Scheme 1 Synthesis of chiral-phosphinoselenoic amide ligands. | ||
Both the enantiomeric pure compounds 1a and 1b show strong absorption at 559 cm−1 in their FT-IR spectrum and can be assigned to the characteristic P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Se bond stretching frequency and it is comparable with the previously observed values: 568 cm−1 for [Ph2P(Se)NHCHPh2], 599 cm−1 for [Ph2P(Se)NHCPh3] and 535 cm−1 for [Ph2P(Se)NHC(CH3)3], which were reported by our group.13 1H NMR spectrum of isomers 1a and 1b shows a doublet resonance signal at δ 1.42 ppm (JH–H = 6.76 Hz) and a multiplet centred at 4.52 ppm respectively, corresponding to the methyl protons and CH proton attached to the α-position of amine nitrogen atom. A broad resonance signal at δ 2.57 ppm represents the amine (N–H) proton of the ligand moiety. These values are observed as slightly downfield shifted when compared to free chiral phosphineamine [Ph2PNH{R-*CHMePh}] or [Ph2PNH{S-*CHMePh}] due to the attachment of the selenium atom to the phosphorous atom.14a
Se bond stretching frequency and it is comparable with the previously observed values: 568 cm−1 for [Ph2P(Se)NHCHPh2], 599 cm−1 for [Ph2P(Se)NHCPh3] and 535 cm−1 for [Ph2P(Se)NHC(CH3)3], which were reported by our group.13 1H NMR spectrum of isomers 1a and 1b shows a doublet resonance signal at δ 1.42 ppm (JH–H = 6.76 Hz) and a multiplet centred at 4.52 ppm respectively, corresponding to the methyl protons and CH proton attached to the α-position of amine nitrogen atom. A broad resonance signal at δ 2.57 ppm represents the amine (N–H) proton of the ligand moiety. These values are observed as slightly downfield shifted when compared to free chiral phosphineamine [Ph2PNH{R-*CHMePh}] or [Ph2PNH{S-*CHMePh}] due to the attachment of the selenium atom to the phosphorous atom.14a
The solid-state structures of 1a and 1b were confirmed using single-crystal X-ray diffraction analysis. The details of the structural parameters are given in Table TS1 in ESI.† The solid-state structures of both enantiomers and selected bond lengths and bond angles are shown in Fig. 1. From the molecular structure of two compounds, it is clear that both enantiomers are non-super imposable mirror images and crystallise in the triclinic space group P1, with one molecule in the unit cell. The PSe bond distances, 2.1219(15) Å (for 1a) and 2.126(2) Å (for 1b), are in good agreement with our previously reported values: 2.1019(8) Å for [Ph2P(Se)NH(2,6-Me2C6H4)],13a 2.1086(12) Å for [Ph2P(Se)NHCHPh2],13c 2.1166(8) Å for [Ph2P(Se)NHCPh3]13c and 2.1187(8) Å for [Ph2P(Se)NHC(CH3)3].13g P1–N1 distance [1.671(5) for 1a and 1.645(5) Å for 1b] and C1–N1 distance [1.454 (7) for 1a and 1.470(9) Å for 1b] are also similar to those of phosphinoselenoic amides [Ph2P(Se)NHR]: P1–N1 1.656(3) Å, C1–N1 1.441(4) Å for R = 2,6-Me2C6H4, P1–N1 1.642(4) Å, C1–N1 1.459 (6) Å for R = CHPh2, P1–N1 1.664(2) Å, C1–N1 1.496(4) Å for R = CPh3 and P1–N1 1.655(3) Å, C1–N1 1.494(4) Å.
 | ||
| Fig. 1 Solid-state structures of ligands 1a (left) and 1b (right). Selected bond lengths (Å) and bond angles (°). | ||
Potassium complexes
The potassium salts of molecular composition [K{N(R-*CHMePh)(Ph2P(Se))}{THF}n] (2a) and [K{N(S-*CHMePh)(Ph2P(Se))}{THF}n] (2b) were readily prepared by the reaction of compound 1a or 1b with potassium precursor [KN(SiMe3)2] in THF via the elimination of volatile bis(trimethylsilyl)amine (see Scheme 2).17 The potassium complexes 2a and 2b were characterised using spectroscopic and analytical techniques. However, suitable crystals for X-ray diffraction analysis were not obtained due to high solubility of the compounds (2a,b) in the THF solvent. In FT-IR spectra, the compound (2a,b) showed a strong absorption band at 570 cm−1 which can be best assigned to characteristic PSe bond stretching and it is in good agreement with our previously described potassium salts of phosphinoselenoic amides: 569 cm−1 for [{(THF)2KPh2P(Se)-N(CHPh2)}2] and 570 cm−1 for [K(THF)2{Ph2P(Se)-N(CMe3)}]n.13c,g 31P{1H} NMR spectra of compound (2a,b) showed a sharp singlet resonance signal at δ 48.6 ppm, which is up-field shifted (56.1 ppm) compared to that of the free ligand (1a,b), indicating clear evidence of the formation of potassium salt. Two multiplet signals in the region of 3.50–3.53 ppm and 1.37–1.40 ppm in 1H spectra also confirm the presence of coordinated THF molecules in the complex 2a,b and using integration it was calculated that three THF molecules were coordinated. One set of signals was observed for the compound (2a,b) in the 1H and 13C{1H} NMR spectra, similar to the free ligand, indicating a dynamic behaviour of the complexes in the solution state.
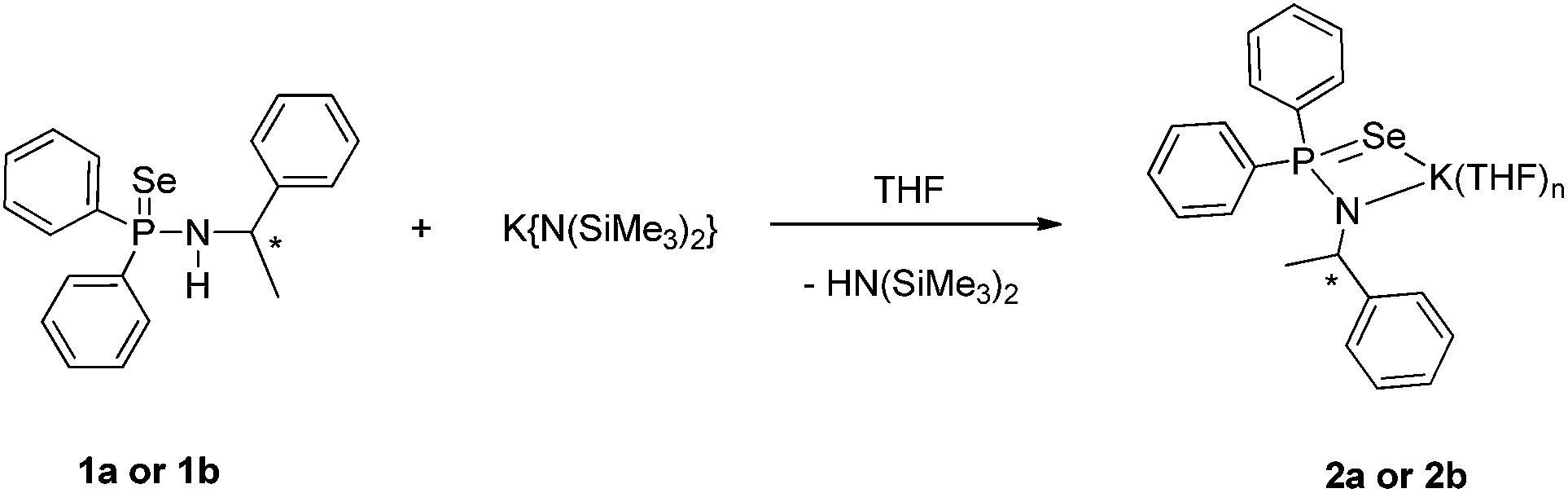 | ||
| Scheme 2 Synthesis of potassium salts 2a and 2b of chiral phosphinoselenoic amides. | ||
Chiral alkaline earth metal complexes
The alkaline earth metal complexes of composition [κ2-{Ph2P(Se)N(R-*CHMePh)}2M(THF)2] (M = Mg (3a), Ca (4a), Sr (5a) and Ba (6a)) and [κ2-{Ph2P(Se)N(S-*CHMePh)}2] [M = Mg (3b), Ca (4b), Sr (5b) and Ba (6b)] were prepared as pure enantiomers by two synthetic methods. In the first method, ligands 1a or 1b were treated with corresponding alkaline earth metal bis(trimethylsilyl)amides [M{N(SiMe3)2}2(THF)n] (M = Ca, Sr and Ba) in 2:1 molar ratio at ambient temperature in THF to afford the desired complexes 4–6 via the elimination of volatile trimethylsilylamine (see Scheme 3).17 In the second, a salt metathesis reaction was employed in which alkaline earth metal diiodies MI2 (M = Mg, Ca, Sr and Ba) were charged with potassium salt [K{N(R-*CHMePh)(Ph2P(Se))}{THF}n] (2a) or [K{N(S-*CHMePh)(Ph2P(Se))}{THF}n] (2b) in 1:2 molar ratio at ambient temperature in THF (see Scheme 3) through the elimination of insoluble potassium iodide. Both methods were used to isolate calcium, strontium and barium complexes. However, the magnesium complexes 3a and 3b were obtained through the second method only. All the complexes are soluble in polar solvents such as THF and dioxan and insoluble in hydrocarbon solvents such as pentane and hexane. Complexes 3a,b–6a,b were re-crystallised through slow evaporation from a THF/n-pentane solution (1:2) at −35 °C. All the complexes were fully characterised using standard analytical/spectroscopic techniques and the solid-state structures of 4a,b–6a,b were confirmed using single-crystal X-ray diffraction analysis.
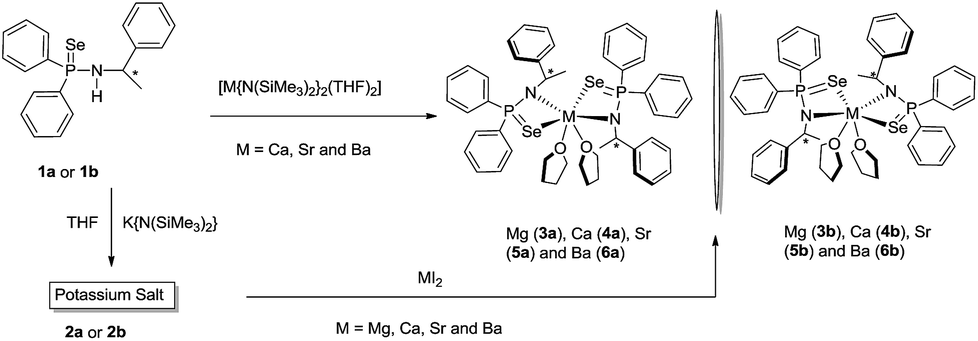 | ||
| Scheme 3 Synthesis of alkaline earth metal complexes of chiral phosphinoselenoic amides. | ||
A strong absorption at 562 cm−1 (for 3a,b), 559 cm−1 for (4a,b), 552 cm−1 (for 5a,b) and 553 cm−1 (for 6a,b) in FT-IR spectra indicates the presence of PSe bond in each metal complex. In 1H NMR spectra, the resonance of one methine proton (CH) α to amido nitrogen was observed as multiplets (δ 4.58–4.62 ppm for 3a,b, 4.26–4.34 ppm for 4a,b, 4.47–4.55 ppm for 5a,b and 4.21–4.29 ppm for 6a,b), which are almost uninfluenced compared to those of free ligands 1a and 1b (4.52 ppm). Doublet signals centred at δ 1.86 (3a,b), 1.68 (4a,b), 1.20 (5a,b) and 1.48 ppm (6a,b) were noticed with coupling constants in the range JH–H = 6.20–6.85 Hz, which can be assigned to methyl protons (–CH3) group attached to the chiral carbon atom in each complex. In 31P{1H} NMR spectra, the magnesium complexes 3a,b showed a sharp resonance signal at δ 45.1 ppm, which is upfield shifted compared to free ligands 1a and 1b. In contrast, the heavier alkaline earth metal complexes 4a,b–6a,b displayed one singlet resonance signal at δ 68.9 ppm, which is significantly downfield shifted compared to that of free ligands 1a and 1b (56.1 ppm) upon coordination of calcium, strontium or barium ion onto the ligand fragment 1. This difference can be attributed to the lower charge of heavier alkaline earth metals, which subsequently causes deshielding of the magnetic field in comparison with the magnesium ion. Similar observations were made in our previously reported complex [(THF)3M{Ph2P(Se)NCH2CH2NPPh2(Se)}] [M = Ca, Sr, Ba] (δ 71–73 ppm) with respect to the magnesium complex [(THF)3Mg{Ph2P(Se)-NCH2CH2NPPh2(Se)}] (43.7 ppm).13f Both the phosphorus atoms present in the ligand moieties {Ph2P(Se)N(*CHMePh)}− are chemically equivalent.
The complexes 3a,b–6a,b represent, to the best of our knowledge, the first alkaline earth metal complexes having chiral phosphinoselenoic amides in the coordination sphere. Thus, molecular structure determinations of these complexes were performed by single-crystal X-ray diffraction techniques. The small crystals obtained from THF/pentane solution of complex 3a,b were found weakly diffracting; however, the solid-state structures of other complexes 4a,b–6a,b confirmed the bi-dentate coordination of the chiral ligand phosphinoselenoic amide. The details of the structural parameters are given in Table TS1 in the ESI.† As a result of the similar ionic radii of the alkaline earth metal ions, the solid-state structures of compounds 4a–6a are isostructural, whereas 4b–6b form the corresponding enantiomers. All the six compounds crystallise in the monoclinic space group P21, with two molecules in the unit cell. Fig. 2 and 3 display the molecular structures of the calcium and barium complexes respectively. Fig. S1 in the ESI† represents the corresponding strontium complexes. In the calcium complexes 4a and 4b, the central calcium atom in each case adopts a distorted octahedral geometry due to κ2-coordination from two ligand moieties and two THF molecules. Each chiral ligand fragment {N(R-*CHMePh)P(Se)Ph2}− and {N(S-*CHMePh)P(Se)Ph2}− is bonded through the amido nitrogen atom and one selenium atom. The Ca–N distances [2.441(3) and 2.426(3) Å] for 4a and [2.444(5) and 2.430(5) Å] for 4b are in good agreement with our structurally characterised calcium complexes: 2.479(5) Å for [Ca{Ph2P(Se)NCHPh2}2(THF)2],13c 2.4534(14) Å for [Ca{Ph2P(BH3)N-CHPh2}2(THF)2],13d 2.386(8) Å for [Ca{C2H4(NPh2PSe)2}(THF)3]13f and 2.451(3) Å for [Ca{Ph2P(Se)NC(CH3)3}2(THF)2].13g However, the observed calcium–nitrogen bond distances are slightly elongated compared to the calcium–nitrogen covalent bond [2.361(2) and 2.335(2) Å] reported for [Ca(Dipp2DAD)(THF)4] (Dipp2DAD) = N,N′-bis(2,6-diisopropylphenyl)-1,4-diaza-1,3-butadiene in the literature.18 The observed Ca–Se bond distances of 3.0303(9) and 3.0794(9) Å for 4a and 3.0327(15) and 3.0817(14) Å for 4b are slightly elongated but within the range of the reported Ca–Se distance of 2.9889(8) Å for structurally characterised complex [Ca{Ph2P(Se)NCHPh2}2(THF)2]13c and 2.9619(3) Å for the complex [Ca{Ph2P(Se)NC(CH3)3}(THF)2]13g and 3.252(2) Å for the complex [Ca{C2H4(NPh2PSe)2}(THF)3].13d The literature reported 2.945(1) Å for [(THF)2Ca{(PyCH)(Se)PPh2}2],19 2.93 Å to 3.00 Å reported for [(THF)4Ca(SeMes′)2] and 2.958(2) Å to 3.001(2) Å reported for [(THF)2Ca(Se2PPh2)2].20,21 The considerably elongated Ca–P distance of 3.2960(13), 3.3013(11) Å in 4a and 3.295(2), 3.3069(19) Å in 4b, was greater than the sum of the covalent radii of calcium and phosphorus (3.07 Å), indicating no interaction between calcium and phosphorus atoms. The PSe distances [2.1444(10), 2.1389(10) Å for 4a and 2.1443(17), 2.1420(16) Å for 4b] are slightly elongated but within the same range as that of the free ligand 1a [2.1219(15) Å]. The P–N distances [1.603(3), 1.607(3) Å for 4a and 1.611(5), 1.602(5) Å for 4b] are slightly shortened compared to the free ligand 1a [1.671(5) Å]. The central calcium atom is additionally ligated by two THF molecules with a Ca–O distance of 2.400(3), 2.444(3) Å for 4a and 2.440(5), 2.394(5) Å for 4b to adopt the calcium atom distorted octahedron geometry. Thus two four-membered metallacycles Ca1–Se1–P1–N1 and Ca1–Se2–P2–N2 are formed due to the ligation of two ligand moieties via selenium and amide nitrogen atoms. The plane containing N1, P1, Se1 and Ca1 makes a dihedral angle of 82.02° (for 4a) and 82.38° (for 4b) with the plane having N2, P2, Se2 and Ca1, indicating that two four-membered metallacycles are almost perpendicular to each other. The O1–Ca1–O2 bond angle is found to be 79.03(13)° for 4a and 78.8(2)° for 4b. Thus the enantiomeric pure compounds 4a and 4b are seen to be fully structurally characterised calcium complexes and, to the best of our knowledge, these are the first examples of chiral calcium complexes with a calcium–selenium direct bond.
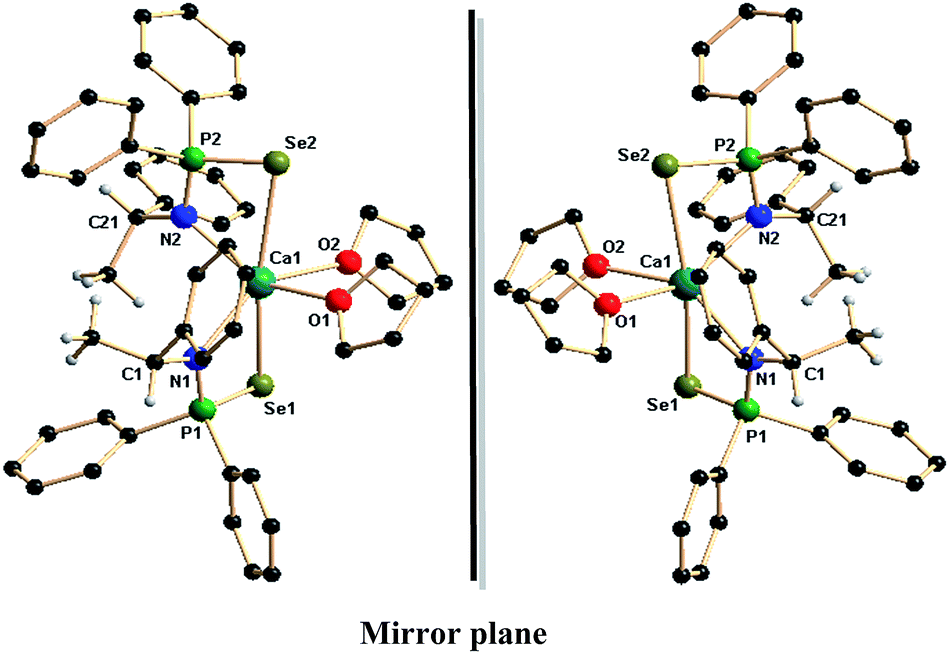 | ||
| Fig. 2 Solid-state structures of calcium complexes 4a and 4b. Hydrogen atoms are omitted for clarity except methyl and methine hydrogen atoms. Selected bond lengths (Å) and bond angles (°). | ||
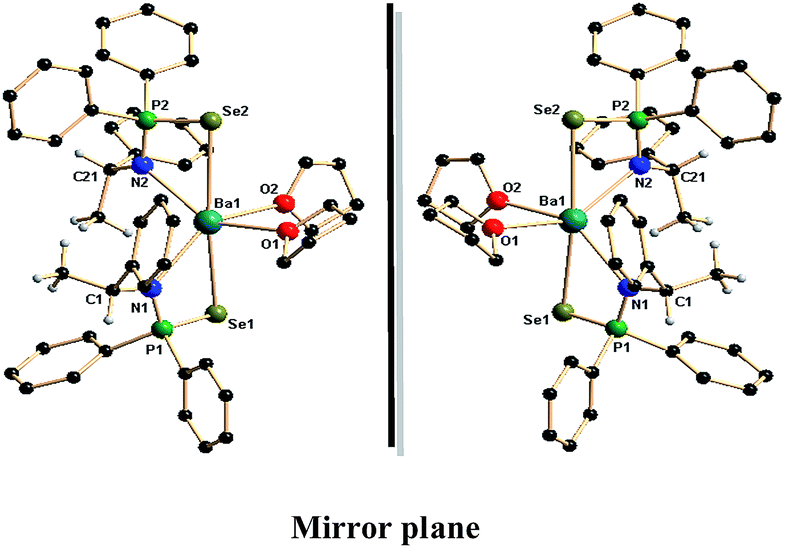 | ||
| Fig. 3 Solid-state structures of barium complexes 6a and 6b. Hydrogen atoms are omitted for clarity except methyl and methine hydrogen atoms. Selected bond lengths (Å) and bond angles (°). | ||
The strontium complex 5a is isostructural to calcium complex 4a due to similar ionic radii of the metal centres (Ca2+ = 1.00 Å; Sr2+ = 1.18 Å for CN = 6)22 and the strontium complex 4b forms the corresponding enantiomer (Fig. S1 in ESI†). In the enantiomeric pure strontium complexes 5a and 5b the strontium ion is six-fold coordinated by the two mono-anionic {N(*CHMePh)P(Se)Ph2}− ligands and two THF molecules. Each ligand {N(*CHMePh)P(Se)Ph2}− coordinates in κ2 fashion via the amido nitrogen atom and one selenium atom to adopt a distorted octahedral geometry for the strontium ion. The Sr–N distances [(2.570(7) and 2.542(7) Å) for 5a and (2.564(5) and 2.569(5) Å) for 5b] fit well with our previously reported strontium–nitrogen bond distances: 2.609(3) Å for the complex [Sr{Ph2P(Se)NCHPh2}2(THF)2] and 2.591(4) Å for [Sr{Ph2P-(BH3)NCHPh2}2(THF)2] and 2.540(5) Å for [Sr{C2H4-(NPh2PSe)2}(THF)3].13 The Sr–Se bond distances of 3.1726(10) and 3.2141(10) Å for 5a, 3.2151(8) and 3.1722(9) Å for 5b are observed, which are quite long, compared to the calcium analogue [3.0327(15) to 3.0817(14) Å] due to the larger ionic radius of the Sr2+ ion. The observed Sr–Se distances in compounds 5a and 5b are within the range of Sr–Se distances [3.138(7) to 3.196(9) Å] of structurally characterised complex [(THF)3Sr(Se2PPh2)2] published by Westerhausen and coworkers23b and 3.066(1) Å for the complex [Sr{Se(2,4,6-tBu3C6H2)}2(THF)4]23a and 3.1356(9) Å for the complex [Sr{Ph2P(Se)NCHPh2}2(THF)2] and 3.2788(10) Å for the complex [Sr{C2H4(NPh2PSe)2}(THF)3] reported by us.13c,d No interaction was observed between the strontium ion and phosphorus atom as the Sr–P distances of 3.449(2), 3.4586(19) Å in 5a and 3.4520(17), 3.4539(15) Å in 5b are greater than the sum of the covalent radii (3.25 Å). Two four-membered metallacycles Sr1–Se1–P1–N1 and Sr1–Se2–P2–N2 are formed due to the ligation of two ligand moieties via selenium and amido nitrogen atoms. The plane containing N1, P1, Se1 and Sr1 makes a dihedral angle of 81.13° (for 5a) and 81.14° (for 5b) against the plane with N2, P2, Se2 and Sr1, indicating that two four-membered metallacycles are almost perpendicular to each other as we observed in the case of calcium complexes (4a and 4b). Thus the enantiomeric pure compounds 5a and 5b are, to the best of our knowledge, the first examples of chiral strontium complexes with a strontium–selenium direct bond.
Similar to calcium and strontium complexes (4a,b–5a,b), the analogous chiral barium complexes 6a and 6b were also crystallised in the monoclinic space group P21 with two molecules in the unit cell. The coordination sphere of the central barium ion of each enantiomer was occupied by two monoanionic {N(*CHMePh)P(Se)Ph2}− ligand moieties where each ligand bonded via amido nitrogen and one selenium atom and two THF molecules through oxygen atoms. Therefore, the central Ba2+ ion in the each enantiomer is six-fold coordinated and adopts distorted octahedral geometry. The Ba–N bond distances of 2.693(4) and 2.679(5) Å for 6a and 2.693(6) and 2.672(7) Å for 6b were observed, which are quite long when compared to analogous calcium [2.441(3) to 2.430(5) Å] and strontium [2.542(7) to 2.570(7) Å] complexes. The observed Ba–N distances are similar to our previously reported values, 2.777(6) and 2.778(6) Å for [Ba{Ph2P(Se)NCHPh2}2(THF)2], 2.733(6) Å for [Ba{Ph2P(BH3)NCHPh2}2(THF)2], (2.657(5) and 2.654(6) Å) for [Ba{C2H4(NPh2PSe)2}(THF)3], and 2.774(5) Å, 2.790(5) and 2.789(5) Å for polymeric ‘ate’ complex of [K(THF)Ba{Ph2P(Se)N(CMe3)}3]n reported by us13 and 2.706(4) Å for [Ba((Dip)2DAD)(μ-I)(THF)2]2 reported in the literature.18 The Ba–Se bond distances of 3.3181(6) and 3.3524(7) Å for 6a and 3.3172(9) and 3.3537(9) Å for 6b were observed and these are within the range of the Ba–Se distances [3.366(1) Å and 3.324(1) Å] for the complex [{BaI(4,5-(P(Se)Ph2)2tz)}2(THF)7] reported by Raymundo Cea-Olivares et al.,24a 3.2787(11) Å for [Ba(THF)4(SeMes*)2] (Mes* = 2,4,6-tBu3C6H2) and 3.2973(3) Å for [{Ba(Py)3(THF)(SeTrip)2}2] (Trip = 2,4,6-iPr3C6H2) reported by Ruhlandt-Senge et al.,24b [3.3553(10) and 3.3314(10) Å] for [Ba{Ph2P(Se)NCHPh2}2(THF)2], 3.3842(8)Å for [{η2-N(PPh2Se)2}2Ba(THF)3] and [3.3274(7) Å, 3.3203(7) Å and 3.3518(7) Å] for [K(THF)Ba{Ph2P(Se)N(CMe3)}3]n previously reported by us.13 The much elongated Ba–P distances of 3.5838(13), 3.5959(15) Å in 6a and 3.5837(18), 3.595(2) Å in 6b indicate no ligation through phosphorus atom to the barium ion. Similar to the calcium and strontium complexes, in 6a and 6b, two four-membered metallacycles Ba1–Se1–P1–N1–Ba1 and Ba1–Se2–P2–N2–Ba1 are formed due to κ2-ligation of two ligand moieties via selenium and amide nitrogen atoms. The plane containing N1, P1, Se1 and Ba1 makes a dihedral angle of 81.37° (for 6a) and 81.29° (for 6b) with the plane having N2, P2, Se2 and Ba1, indicating that the two four-membered metallacycles are almost perpendicular to each other as we observed in the case of the calcium (4a and 4b) and strontium (5a and 5b) complexes. Thus, the enantiomeric pure compounds 6a and 6b are seen to be new class of alkaline earth metal molecules and to the best of our knowledge; these are the first examples of chiral barium complexes with a barium–selenium direct bond.
Chiral amidophosphine-borane ligands
In our previous studies on phosphine-borane adducts in alkali and alkaline earth metal chemistry, we introduced a monoanionic amidophosphine-borane {Ph2P(BH3)NR}− (R = CHPh2 and CPh3) and dianionic bis(amidodiphenylphosphine-borane) {Ph2P(BH3)NCH2CH2NP(BH3)Ph2}2− as chelating ligands and exploited their chelating behaviour in alkali metal and alkaline earth metal chemistry.13d,f The monoanionic amidophosphine-borane {Ph2P(BH3)NR}− acts as bi-dentate ligand and coordinates to the metal ions through amido nitrogen and borane hydrogens, whereas bis(amido-diphenylphosphine-borane) would form a dianion and acts as a tetra-dentate ligand towards metal ions. To extend our research on amidophosphine-boranes and demonstrate the versatility of the amidophosphine-boranes mainly in alkaline earth metal chemistry, we intended to develop chiral amidophosphine-borane ligands [HN(R-*CHMePh)(P(BH3)Ph2)] (7a) and [HN(S-*CHMePh)-(P(BH3)Ph2)] (7b) and their homoleptic barium complexes [Ba{N(R-*CHMePh)P(BH3)Ph2}2(THF)2] (8a) and [Ba{N(R-*CHMePh)P(BH3)Ph2}2(THF)2] (8b). The amido-phosphineborane (7a) and (7b) were isolated as pure enantiomers from a single-step reaction involving corresponding chiral phosphineamines [HN(R-*CHMePh)(PPh2)] and [HN(S-*CHMePh)(PPh2)] and borane adduct [H3B·SMe2] at room temperature in a 1:1 molar ratio in toluene (see Scheme 4).17
 | ||
| Scheme 4 Synthesis of chiral amidophosphine-borane ligands 7a and 7b. | ||
The formation of the chiral amidophosphine-borane ligands 7a and 7b from [HN(R-*CHMePh)(PPh2)] and [HN(S-*CHMePh)(PPh2)] can easily be followed by 1H NMR spectroscopy measured in CDCl3, since additional resonances for the two chemically equivalent borane (BH3) groups attached to the phosphorus atoms appear as a broad signal centered at δ 0.96 ppm. In the 1H NMR spectra, the resonance signals of ligands 7a,b are marginally shifted in comparison to the starting material with those reported for the phosphineamines.14a The multiplet signals at δ 4.47–4.37 ppm can be assigned to the methine proton (–CH) α to amino nitrogen of ligand 7a,b. A broad signal centered at δ 2.48 ppm corresponding to the –NH proton of ligand 7a,b was observed and also downfield shifted (3.24 ppm) compared to that in 1a,b (2.57 ppm). Ligands 7a,b show a doublet signal at δ 1.40 ppm with coupling constant of JH–H = 6.76 Hz, corresponding to the methyl (–CH3) protons of the ligand 7a,b. In the 31P{1H} NMR spectra, the doublet resonance signal at δ 54.9 ppm with a coupling constant of JP–B = 80.95 Hz can be attributed to the coupling of the phosphorus atom with the adjacent boron atom. In the 11B{1H} NMR spectrum, the broad signal at −37.9 ppm can be assigned to the BH3 group attached to the phosphorus atom. This observation is in agreement with our previously reported values.13d In the FT-IR spectra, a characteristic signal for P–B bond stretching at 608 cm−1 was observed along with another characteristic signal at 2379 cm−1 assigned to the B–H stretching frequency. These values are in agreement with those reported in literature.
The molecular structures of enantiomers 7a and 7b were established using single-crystal X-ray diffraction analysis. R-isomer (7a) crystallises in the monoclinic space group P21, with two independent molecules in the unit cell, whereas the corresponding S-isomer (7b) crystallises in the orthorhombic space group P212121 with eight independent molecules in the unit cell. The details of the structural parameters are given in Table TS1 in ESI.† Fig. 4 represents the molecular structure of 7a and 7b. The P1–B1 bond distances [1.915(5) Å (7a) and 1.892(3) Å (7b)] are almost similar and in full agreement with reported values, 1.918(6) Å for [Ph2P(BH3)-NH(CHPh2)], 1.9091(2) and 1.916(1) Å for [{Ph2P(BH3)NH-CH2-CH2-NHP(BH3)Ph2] and 2.1019(8) Å for [{Ph2P(BH3)}2CH2] and 1.921(3) Å for [(CH2-o-CF3C6H4)-(Ph)P(BH3)C4H8P(BH3)(Ph)-(CH2-o-CF3C6H4)], so they may be considered as the phosphorus–boron dative bond reported by us and others.25 The P1–N1 bond ranges from 1.638(3) Å to 1.653(2) Å and C1–N1 bond distances of 1.466(4) Å and 1.478(3) are also similar to those reported by us previously:13 P1–N1 1.673(6) Å and C1–N1 1.453(8) Å for [Ph2PNH(CHPh2)] and P1–N1 1.638(3) Å and C1–N1 1.468(5) Å for [Ph2P(BH3)NH(CHPh2)].
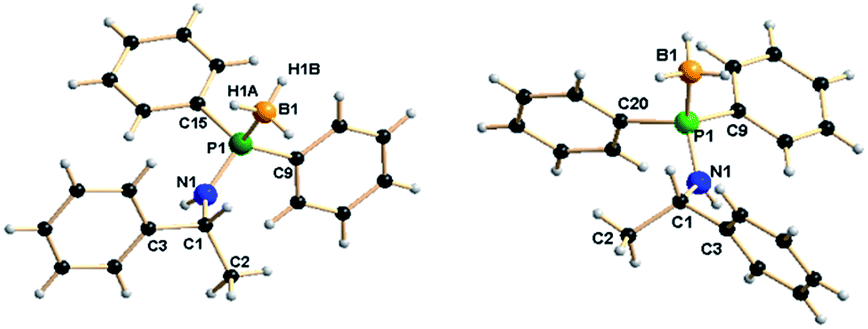 | ||
| Fig. 4 Solid-state structures of two enantiomers 7a (left) and 7b (right). Selected bond lengths (Å) and bond angles (°). | ||
Barium complexes
In a single pot-reaction, chiral ligands 7a or 7b was made to react with [K{N(SiMe3)2}] in THF at an ambient temperature in a 1:1 molar ratio followed by the addition of barium diiodide to afford the barium complexes [(THF)2Ba{N(R-*CHMePh)(P(BH3)Ph2)}2] (8a) and [(THF)2Ba{N(S-*CHMePh)(P(BH3)Ph2)}2] (8b) through the elimination of KI and volatile [HN(SiMe3)2] (see Scheme 5).17
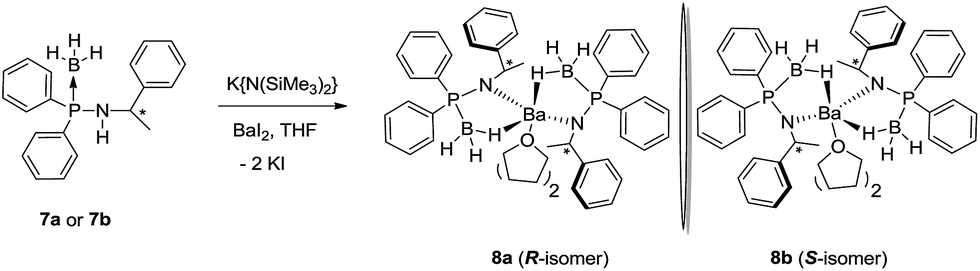 | ||
| Scheme 5 Synthesis of barium complexes 8a and 8b of chiral amidophosphine-borane ligands. | ||
In FT-IR spectra, a strong absorption band at 602 cm−1 is assigned to the P–B bond of complexes 8a,b which is in the range similar to that of ligand 7a or 7b (608 cm−1). The 1H NMR spectra of complex 8a,b measured in C6D6 are very similar to the spectra recorded for ligand 7a or 7b and reveal time-averaged Cs-symmetry in solution. Methyl protons in the ligand backbone appear as a doublet at δ 1.40 ppm with a coupling constant of 6.76 Hz. The resonances of the three protons attached to the boron atom appear as multiplets centered at δ 1.22 ppm in the 1H NMR spectra. Methine proton of the anionic ligand in the barium complexes 8a,b observed as multiplet signals in the region of δ 4.39–4.48 in the 1H NMR spectra. In the proton decoupled 31P NMR spectra, complexes 8a,b show only one doublet signal at δ 46.9 ppm and this value is significantly up-field shifted compared to the value obtained for compound 7a or 7b (54.9 ppm) upon the coordination of barium ion to the ligand 7a or 7b. The phosphorus atoms present in the [N(*CHMePh)P(BH3)Ph2]− moieties are chemically equivalent. A broad signal centered at δ −34.9 ppm was observed in the 11B{1H} NMR spectra of complexes 8a,b.
Although there is ongoing interest in alkaline earth organometallics26 and particularly in the cyclopentadienyl chemistry of these elements,27 complexes 8a,b represent, to the best of our knowledge, the first barium complexes containing a chiral amidophosphine-borane ligand in its coordination sphere. Therefore, the molecular structure in the solid state was determined using X-ray diffraction analysis. Compounds 8a and 8b were re-crystallised by slow evaporation from THF and n-pentane mixture (1:2) and was found to crystallise in the monoclinic space group P21 with two molecules in the unit cell. The solid-state structures of complexes 8a,b confirmed the attachment of chiral amidophosphine-borane ligand onto the barium ion. Fig. 5 shows the non-super imposable mirror images of barium complexes 8a and 8b. The details of the structural parameters are given in Table TS1 in the ESI.† The enantiomeric pure barium compounds 8a,b are non-centrosymmetric and each barium ion in 8a and 8b is coordinated by two amido nitrogen atoms and two BH3 groups of two ligand fragments. One of the borane (BH3) groups coordinates through the hydrogen atoms in a η1 fashion and has a Ba1–B1 distance of 3.221(6) Å. The second borane (BH3) group coordinates in η2 fashion and has Ba1–B2 distance of 3.155(6) Å. Thus, ligand 7a or 7b can be considered a pseudo bi-dentate ligand, similar to {Ph2P(BH3)N(CHPh2)} which was previously introduced into alkaline earth metal chemistry by us.13c Additionally, two THF molecules are coordinated to each barium ion and the geometry around each barium ion is best described as a distorted octahedral. It must be noted that the P–B distances [1.929(6) and 1.924(6) Å] are in the same range as that of the ligands 7a [1.915(5) Å] and 7b [1.892(3) Å] even after the ligation of the BH3 group to the barium centre. The Ba–N [2.674(4), 2.684(4) Å] and Ba1–O1 [2.759(4) and 2.697(4) Å] distances are in agreement with those of the reported complexes.28
 | ||
| Fig. 5 Solid-state structures of barium complexes 8a and 8b. Hydrogen atoms are omitted for clarity except for methyl, methine as well as for borane hydrogen atoms. Selected bond lengths (Å) and bond angles (°). | ||
Ring-opening polymerisation study
Catalytic activities of the chiral strontium and barium complexes 5a or 5b and 6a or 6b were performed (see Scheme 6). Polymerisation studies were typically conducted in toluene, with various monomer/catalyst ratios at 25 °C. Selected data obtained with respect to complexes 5 and 6 are given in Table 1. | ||
| Scheme 6 Ring-opening polymerisation of ε-CL with strontium and barium complexes 5 and 6. | ||
| Entry | [M] | [ε-CL]0/[M]0 | Reac. timeb [min] | Conv.c [%] | Mn (theo)d [g mol−1] | Mn (GPC)e [g mol−1] | Mw (GPC)e [g mol−1] | Mw/Mn (PDI)f |
|---|---|---|---|---|---|---|---|---|
| a Results are representative of at least two experiments.b Reaction times were not necessarily optimized.c Monomer conversions were determined by 1H NMR spectroscopy.d Theoretical molar mass values calculated from the relation: [monomer]0/[M]0 × monomer conversion where [M]0 = 8.76 × 10−3 mmol and monomer weight of ε-CL = 114 g mol−1.e Experimental molar masses were determined by GPC versus polyethylene glycol standards.f Molar mass distributions were calculated from GPC. | ||||||||
| 1 | Sr | 100 | 15 | 90 | 9001 | 8797 | 17108 |
1.94 |
| 2 | Sr | 200 | 15 | 80 | 16603 |
10515 |
17153 |
1.63 |
| 3 | Sr | 300 | 15 | 73 | 21904 |
12717 |
19707 |
1.54 |
| 4 | Sr | 400 | 15 | 82 | 32807 |
20492 |
32065 |
1.56 |
| 5 | Sr | 500 | 15 | 75 | 37508 |
22261 |
24295 |
1.09 |
| 6 | Ba | 100 | 10 | 98 | 9802 | 8829 | 11512 |
1.30 |
| 7 | Ba | 200 | 10 | 90 | 18003 |
10351 |
14450 |
1.39 |
| 8 | Ba | 300 | 10 | 85 | 25505 |
11735 |
17467 |
1.48 |
| 9 | Ba | 400 | 10 | 80 | 32007 |
12620 |
19581 |
1.55 |
| 10 | Ba | 500 | 10 | 83 | 43509 |
32338 |
37336 |
1.15 |
The catalytic ability of the newly synthesised enantiomeric pure mono-nuclear strontium complexes 5a or 5b to promote the ROP of ε-CL was first evaluated (Table 1, entries 1–5). Indeed, the moderate reactivity of the strontium complexes is very similar to that observed in previously reported studies using other strontium complexes for ROP of ε-caprolactone.29 Since the larger ion radius barium complexes have been reported to be more active than the calcium and strontium congeners in ROP,30,31 we tested compound 6a or 6b as a catalyst and observed an enhanced rate of polymerisation (Table 1, entries 6–10). In the case of strontium, higher reactivity was observed for conversion of ε-caprolactone to poly-caprolactone and up to 500 ε-CL units were successfully converted in high yields (75–90 per cent), within 15 and 10 minutes respectively, at 25 °C. The control over the ROP process was rather good, affording PCLs, featuring a considerable match between the observed (as determined by GPC) and calculated molar mass values, as well as moderate dispersity data (PDI = Mw/Mn < 1.94). However, the overall efficiency of the strontium initiator 5a,b towards the ROP of ε-CL was weaker than that of the barium analogue 6a,b. Being the largest ionic radius of the barium atom, it was anticipated that complex 6a,b would show the highest reactivity among all the three alkaline earth metal complexes.32,33 In reality we observed that up to 500 ε-CL units were successfully converted in good yields (80–98 per cent) within 10 minutes at 25 °C (Table 1, entries 6–10). The poly-caprolactone produced by the use of the barium catalyst was a considerable match between the observed and calculated molar mass values, and we observed a relatively narrow poly-dispersity data (PDI up to 1.55, entry 9 in Table 1). Thus, among strontium and barium metal complexes, the barium complexes 6a,b showed the highest activity for ROP of ε-caprolactone.
Experimental
General consideration
All manipulations of air-sensitive materials were performed with the rigorous exclusion of oxygen and moisture in flame-dried Schlenk-type glassware either on a dual manifold Schlenk line, interfaced to a high vacuum (10−4 Torr) line, or in an argon-filled M. Braun glove box. THF was pre-dried over Na wire and distilled under nitrogen from sodium and benzophenone ketyl prior to use. Hydrocarbon solvents (toluene and n-pentane) were distilled under nitrogen from LiAlH4 and stored in the glove box. 1H NMR (400 MHz), 13C{1H} and 31P{1H} NMR (161.9 MHz) spectra were recorded on a BRUKER AVANCE III-400 spectrometer. BRUKER ALPHA FT-IR was used for FT-IR measurement. Elemental analyses were performed on a BRUKER EURO EA at the Indian Institute of Technology Hyderabad. Alkaline earth metal diiodides (MgI2, CaI2, SrI2 and BaI2), KN(SiMe3)2, selenium and Me2S·BH3 were purchased from Sigma Aldrich and used as such. The chiral-aminophosphines [HN(R-*CHMePh)(PPh2)], [HN(S-*CHMePh)(PPh2)] were prepared according to procedure prescribed in the literature.14a The NMR solvent C6D6 and CDCl3 were purchased from Sigma Aldrich and dried under Na/K alloy (for C6D6) or molecular sieves (for CDCl3) prior to use.Preparation of [Ph2P(Se)HN(R-*CHMePh)] (1a) and [Ph2P(Se)HN(S-*CHMePh)] (1b)
In a 25 ml round-bottomed flask, chiral-aminophosphines [HN(R-*CHMePh)(PPh2)] or [HN(S-*CHMePh)(PPh2)] (1.0 g, 3.27 mmol) and elemental selenium (392 mg, 4.91 mmol) were heated to 60 °C in THF (10 ml) solvent for 12 hours. Excess selenium metal was filtered through a G4 frit to collect the yellow-coloured filtrate. After evaporation of the solvent from filtrate in vacuo, a light-yellow solid residue was obtained, which was further purified by washing with n-hexane. Compound 1a was re-crystallised from THF at room temperature.Yield: 1.24 g (98%) (1a) and 1.25 g (99%) (1b). 1H NMR (400 MHz, CDCl3): δ 7.88–7.94 (m, 2H, ArH), 7.71–7.77 (m, 2H, ArH), 7.30–7.39 (m, 4H, ArH), 7.12–7.25 (m, 7H, ArH), 4.43–4.52 (m, 1H, CH), 2.57 (br, 1H, NH), 1.42 (d, JH–H = 6.76 Hz, 3H, CH3) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 144.7 (ArC), 132.0 (P-ArC), 131.8 (P attached o-ArC), 131.6 (o-ArC), 128.5 (P attached m-ArC), 128.3 (m-ArC), 127.1 (p-ArC), 126.3 (P attached p-ArC), 52.7 (CH), 25.2 (CH3) ppm. 31P{1H} NMR (161.9 MHz, CDCl3): δ 56.1 ppm. FT-IR (selected frequencies): ν = 3501 (N–H), 1434 (P–C), 954 (P–N), 556 (PSe) cm−1. Elemental analysis: C20H20NPSe (385.05): calcd C 62.50, H 5.25, N 3.64. Found C 62.28, H 5.13, N 3.42.
Preparation of [K{N(R-*CHMePh)(Ph2P(Se))}(THF)n] (2a) and [K{N(S-*CHMePh)(Ph2P(Se))}(THF)n] (2b)
In a 50 ml pre-dried Schlenk flask, one equivalent (1.00 g, 2.60 mmol) of ligand 1a and one equivalent of potassium bis(trimethylsilyl)amide (520 mg, 2.60 mmol) were mixed together with 10 ml of dry THF. After 6 hours of stirring, the THF solvent was evaporated in vacuo and the dry compound was further purified by washing with n-pentane (5 ml) twice. The title compound 2a was obtained as a light orange powder. Compound 2b was also obtained by similar procedure.Yield: 1.24 g, (90%) (2a) and yield: 1.20 g (86%) (2b). 1H NMR (400 MHz, C6D6): δ 7.92–8.08 (m, 4H, ArH), 7.34 (bs, 2H, ArH), 7.01–7.19 (m, 9H, ArH), 4.31–4.37 (m, 1H, CH), 3.50–3.53 (m, THF), 1.37–1.40 (m, THF), 1.30 (d, JH–H = 6.20 Hz, 3H, CH3) ppm. 13C{1H} NMR (100 MHz, C6D6): δ 144.7 (ArC), 132.0 (P-ArC), 131.8 (P attached o-ArC), 131.6 (o-ArC), 128.7 (P attached m-ArC), 128.4 (m-ArC), 127.7 (p-ArC), 126.7 (P attached p-ArC), 67.6 (THF), 52.7 (CH), 26.1 (CH3), 25.6 (THF) ppm. 31P{1H} NMR (161.9 MHz, C6D6): δ 42.7 ppm. FT-IR (selected frequencies): ν = 1435 (P–C), 955 (P–N), 550 (PSe) cm−1. Elemental analysis: C28H35KNO2PSe (567.12): calcd C 59.35, H 6.23, N 2.47. Found C 58.84, H 5.99, N 2.23.
Preparation of [{(THF)2Mg{Ph2P(Se)N(R-*CHMePh)}2] (3a) and [{(THF)2Mg{Ph2P(Se)N(S-*CHMePh)}2] (3b)
In a 25 ml pre-dried Schlenk flask, potassium salt of ligand 1a (304 mg, 0.72 mmol) (1b for 3b) was mixed with MgI2 (100 mg, 0.36 mmol) in 10 ml THF solvent at ambient temperature and stirring continued for 12 hours. The white precipitate of KI was filtered off and the filtrate was evaporated in vacuo. The resulting white residue was further purified by washing with n-pentane and crystals suitable for X-ray analysis were grown from THF/n-pentane (1:2) mixture at −35 °C.
Yield: 154.0 mg, (90%) (3a) and 125 mg (80%) (3b). 1H NMR (400 MHz, C6D6): δ 8.06–8.12 (m, 1H, ArH), 7.91–7.98 (m, 2H, ArH), 7.76–7.82 (m, 1H, ArH), 7.40–7.42 (m, 1H, ArH), 6.89–7.11 (m, 10H, ArH), 4.58–4.62 (m, 1H, CH), 3.68–3.74 (m, THF), 1.86 (d, JH–H = 6.72 Hz, 3H, CH3), 1.42–1.44 (m, THF) ppm. 13C{1H} NMR (100 MHz, C6D6): δ 144.7 (ArC), 132.0 (P-ArC), 131.8 (P attached o-ArC), 131.6 (o-ArC), 128.5 (P attached m-ArC), 128.3 (m-ArC), 127.1 (p-ArC), 126.3 (P attached p-ArC), 52.7 (CH), 25.2 (CH3) ppm. 31P{1H} NMR (161.9 MHz, C6D6): δ 45.1 ppm. FT-IR (selected frequencies): ν = 1435 (P–C), 955 (P–N), 562 (PSe) cm−1. Elemental analysis: C38H48MgN2O3P2Se2 (826.13): calcd C 55.32, H 5.86, N 3.40. Found C 54.93, H 5.62, N 3.13.
Preparation of [{(THF)2Ca{Ph2P(Se)N(R-*CHMePh)}2] (4a) and [{(THF)2Ca{Ph2P(Se)N(S-*CHMePh)}2] (4b)
:2) mixture at −35 °C.Yield: 154.0 mg, (90%) (4a) and 149 mg (86%) (4b). 1H NMR (400 MHz, C6D6): δ 7.95–8.00 (m, 2H, ArH), 7.63–7.94 (m, 2H, ArH), 7.43–7.45 (m, 2H, ArH), 6.85–7.06 (m, 9H, ArH), 4.26–4.34 (d, 1H, CH), 3.63 (m, THF), 1.68 (d, JH–H = 6.56 Hz, 3H, CH3), 1.21 (m, THF) ppm. 13C{1H} NMR (100 MHz, C6D6): δ 150.2 (ArC), 150.1 (ArC), 132.6 (P-ArC), 132.4 (P attached o-ArC), 130.2 (o-ArC), 129.9 (P attached m-ArC), 128.6 (m-ArC), 126.6 (p-ArC), 125.9 (P attached p-ArC), 69.0 (THF), 58.7 (CH), 30.0 (CH3), 25.5 (THF) ppm. 31P{1H} NMR (161.9 MHz, C6D6): δ 69.8 ppm. FT-IR (selected frequencies): ν = 1435 (P–C), 954 (P–N), 559 (PSe) cm−1. Elemental analysis: C48H54CaN2O2P2Se2 (950.87): calcd C 60.63, H 5.72, N 2.95. Found C 60.41, H 5.66, N 2.86.
Preparation of [{(THF)2Sr{Ph2P(Se)N(R-*CHMePh)}2] (5a) and [{(THF)2Sr{Ph2P(Se)N(S-*CHMePh)}2] (5b)
:2) mixture at −35 °C.Yield: 154.0 mg, (90%) (5a) and 145 mg (85%) (5b). 1H NMR (400 MHz, C6D6): δ 7.98–8.03 (m, 2H, ArH), 7.78–7.84 (m, 4H, ArH), 6.90–6.97 (m, 2H, ArH), 6.78–87 (m, 7H, ArH), 4.47–4.55 (m, 1H, CH), 3.45–3.48 (m, THF), 1.29–1.32 (m, THF), 1.20 (d, JH–H = 6.80 Hz, 3H, CH3) ppm. 13C{1H} NMR (100 MHz, C6D6): δ 145.4 (ArC), 145.3 (ArC), 135.4 (P-ArC), 134.5 (P-ArC), 132.3 (P attached o-ArC), 131.2 (o-ArC), 128.1 (P attached m-ArC), 127.9 (m-ArC), 127.7 (p-ArC), 126.4 (P attached p-ArC), 67.6 (THF), 52.5 (CH), 25.6 (THF), 25.1 (CH3) ppm. 31P{1H} NMR (161.9 MHz, C6D6): δ 69.8 ppm. FT-IR (selected frequencies): ν = 1434 (P–C), 955 (P–N), 552 (PSe) cm−1. Elemental analysis: C48H54N2O2P2Se2Sr (998.41): calcd C 57.74, H 5.45, N 2.81. Found C 57.50, H 5.29, N 2.61.
Preparation of [{(THF)2Ba{Ph2P(Se)N(R-*CHMePh)}2] (6a) and [{(THF)2Ba{Ph2P(Se)N(S-*CHMePh)}2] (6b)
:2) mixture at −35 °C.Yield: 154.0 mg, (90%) (6a) and 156 g, (91%) (6b). 1H NMR (400 MHz, C6D6): δ 7.96–7.99 (m, 2H, ArH), 7.62–7.66 (m, 2H, ArH), 7.19–7.29 (m, 4H, ArH), 6.90–7.06 (m, 7H, ArH), 4.21–4.29 (m, 1H, CH), 3.54–3.57 (m, THF), 1.48 (d, JH–H = 6.20 Hz, 3H, CH3), 1.35–1.38 (m, THF) ppm. 13C{1H} NMR (100 MHz, C6D6): δ 144.7 (ArC), 132.1 (P-ArC), 130.2 (P attached o-ArC), 129.5 (o-ArC), 127.8 (P attached m-ArC), 126.8 (m-ArC), 126.4 (p-ArC), 126.3 (P attached p-ArC), 68.0 (THF), 52.7 (CH), 25.2 (CH3), 25.6 (THF) ppm. 31P{1H} NMR (161.9 MHz, C6D6): δ 69.8 ppm. FT-IR (selected frequencies): ν = 1435 (P–C), 956 (P–N), 553 (PSe) cm−1. Elemental analysis: C48H54BaN2O2P2Se2 (1048.12): calcd C 55.00, H 5.19, N 2.67. Found C 54.81, H 4.91, N 2.42.
Preparation of [Ph2P(BH3)HN(R-*CHMePh)] (7a) and [Ph2P(BH3)HN(S-*CHMePh)] (7b)
In a pre-dried Schlenk flask 1.0 g (3.27 mmol) of chiral-aminophosphines [HN(R-*CHMePh)(PPh2)] or [HN(S-*CHMePh)-(PPh2)] was placed in 10 ml of toluene, and to this solution, borane-dimethyl sulfide (0.30 ml, 3.27 mmol) in 5 ml of toluene was added drop wise with constant stirring at room temperature. The reaction mixture was then stirred for another 12 hours. A white precipitate was formed and was filtered through a G4 frit and dried in vacuo. The pure compound was obtained after washing with n-pentane.Yield: 1.20 g (100%) (7a) and 1.20 g (100%) (7b). Compound 7a was soluble in CDCl3, CH2Cl2, THF, and hot toluene. It was re-crystallised from hot toluene. 1H NMR (400 MHz, CDCl3): δ 7.60–7.54 (m, 4H, ArH), 7.45–7.30 (m, 6H, ArH), 7.24–7.15 (m, 5H, ArH), 4.47–4.37 (m, 1H, CH), 2.48 (br, 1H, NH), 1.40 (d, JH–H = 6.76 Hz, 3H, CH3), 1.17–0.75 (br, 3H, BH3) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 145.1 (ArC), 132.4 (P-ArC), 131.9 (P attached o-ArC), 131.1 (o-ArC), 128.5 (P attached m-ArC), 128.3 (m-ArC), 127.0 (p-ArC), 125.8 (P attached p-ArC), 53.1 (CH), 25.9 (CH3) ppm. 31P{1H} NMR (161.9 MHz, CDCl3): δ 54.9 (d, JP–B = 80.95 Hz) ppm. 11B{1H} NMR (128.4 MHz, CDCl3): δ −37.9 (br) ppm. FT-IR (selected frequencies): ν 3438 (N–H), 1436 (P–C), 909 (P–N), 2379 (B–H), 608 (P–B) cm−1. Elemental analysis: C20H23BNP (319.17): calcd C 75.26, H 7.26, N 4.39. Found C 74.82, H 6.91, N 4.22.
Preparation of [{(THF)2Ba{Ph2P(BH3)N(R-*CHMePh)}2] (8a) and [{(THF)2Ba{Ph2P(BH3)N(S-*CHMePh)}2] (8b)
In a 25 ml pre-dried Schlenk flask, ligand 7, potassium bis(trimethylsilyl)amide and BaI2 (100 mg, 0.26 mmol) were mixed in 10 ml THF solvent at ambient temperature and stirring continued for 12 hours. The white precipitate of KI was filtered off and filtrate was evaporated in vacuo. The resulting white residue was further purified by washing with n-pentane and crystals suitable for X-ray analysis were grown from THF/n-pentane (1:2) mixture at −35 °C.
Yield: 154.0 mg, (90%) (8a) and 156 g, (91%) (8b). 1H NMR (400 MHz, C6D6): δ 7.60–7.54 (m, 4H, ArH), 7.45–7.30 (m, 6H, ArH), 7.247.15 (m, 5H, ArH), 4.48–4.39 (m, 1H, CH), 2.48 (br, 1H, NH), 1.40 (d, JH–H = 6.76 Hz, 3H, CH3), 1.49–0.94 (br, 3H, BH3) ppm. 13C{1H} NMR (100 MHz, C6D6): δ 144.7 (ArC), 132.0 (P-ArC), 131.8 (P attached o-ArC), 131.6 (o-ArC), 128.5 (P attached m-ArC), 128.3 (m-ArC), 127.1 (p-ArC), 126.3 (P attached p-ArC), 52.7 (CH), 25.2 (CH3) ppm. 31P{1H} NMR (161.9 MHz, C6D6): δ 46.9 ppm. 11B{1H} NMR (128.4 MHz, C6D6): δ −34.9 (d) ppm. FT-IR (selected frequencies): ν = 1434 (P–C), 999 (P–N), 2383 (B–H), 602 (P–B) cm−1. Elemental analysis: C48H60B2BaN2O2P2 (917.87): calcd C 62.81, H 6.59, N 3.05. Found C 61.94, H 6.20, N 2.83.
Typical polymerisation experiment
In a glove box under argon atmosphere, the catalyst was dissolved in the appropriate amount (1.0 ml) of dry toluene. ε-Caprolactone in 1.0 ml of toluene was then added along with vigorous stirring. The reaction mixture was stirred at room temperature for 5–20 minutes, after which the reaction mixture was quenched by the addition of a small amount of (1.0 ml) methanol. Later, a small quantity of excess acidified methanol was added. The polymer was precipitated in excess methanol and it was filtered and dried under vacuum. The final polymer was then analysed by NMR and GPC.X-ray crystallographic studies of 1, 4–8
Single crystals of compounds 1a,b were grown from a concentrated solution of THF at room temperature. However, the single crystals of 4a,b–8a,b suitable for X-ray measurement were grown at −35 °C under inert atmosphere. For compounds 4a,b–8a,b, (except 7a,b) a crystal of suitable dimensions was mounted on a CryoLoop (Hampton Research Corp.) with a layer of light mineral oil and placed in a nitrogen stream at 150(2) K. However for compounds 1a,b and 7a,b, the data were collected at 293 K. All measurements were made on an agillent Supernova X-calibur Eos CCD detector with graphite-monochromatic Cu-Kα (1.54184 Å) radiation. Crystal data and structure refinement parameters are summarised in Table TS1 in the ESI.† The structures were solved by direct methods (SIR92)34 and refined on F2 by full-matrix least-squares methods; using SHELXL-97.35 Non-hydrogen atoms were anisotropically refined. H atoms were included in the refinement in calculated positions riding on their carrier atoms. No restraint was made with respect to any of the compounds. The function minimised was [∑w(Fo2 − Fc2)2] (w = 1/[σ2(Fo2) + (aP)2 + bP]), where P = (max(Fo2,0) + 2Fc2)/3 with σ2(Fo2) from counting statistics. The function R1 and wR2 were (∑‖Fo| − |Fc‖)/∑|Fo| and [∑w(Fo2 − Fc2)2/∑(wFo4)]1/2, respectively. The Diamond-3 program was used to draw the molecule.†Conclusion
We have demonstrated a series of alkaline earth metal complexes obtained in enantiomeric pure form with chiral phosphinoselenoic amides ligand through two routes of synthesis. In the solid-state structures of Ca–Ba complexes, the monoanionic ligand attached to the metal centre in κ2 fashion via the coordination of amido nitrogen and selenium atoms, confirming the bidentate chelation of chiral phosphinoselenoic amide. Thus, the enantiomeric pure compounds 4–6 are known to be a new class of alkaline earth metal complexes, and to the best of our knowledge, these are the first examples of chiral alkaline earth metal complexes with a metal-selenium direct bond. We have also described the synthetic and structural features of chiral amidophosphine-borane ligands and the corresponding barium complex. It was found that the amidophosphine-borane ligand is coordinated through the amido nitrogen and BH3 hydrogens (η1 and η2) to the barium ion. We have tested complexes 5–6 as catalysts for the ROP of ε-caprolactone and observed that the barium complex, having the largest ionic radius, acts as the best catalyst between the two analogous complexes.Acknowledgements
This work was supported by the Department of Science and Technology India (DST) under the SERC Fast Track Scheme (SR/FT/CS-74/2010) and start-up grant from IIT Hyderabad. R. K. Kottalanka thanks the University Grants Commission, A. Harinath thanks the Council of Scientific & Industrial Research (CSIR) India for their PhD fellowship. We thank Prof. K. Mashima, Division of Chemistry, Graduate School of Engineering Science, Osaka University for providing the facility to measure the polymers.Notes and references
- (a) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, USA, 1998 Search PubMed; (b) B. M. Trost, Science, 1991, 254, 1471 CAS; (c) B. M. Trost, Angew. Chem., Int. Ed., 1995, 34, 259 CrossRef CAS PubMed; (d) P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686 CrossRef CAS PubMed.
- (a) Y. Yamashita, T. Tsubogo and S. Kobayashi, Chem. Sci., 2012, 3, 967–975 RSC; (b) S. Kobayashi and Y. Yamashita, Acc. Chem. Res., 2011, 44, 58–71 CrossRef CAS PubMed; (c) A. Yanagisawa and K. Yoshida, Synlett, 2011, 20, 2929–2938 CrossRef PubMed; (d) S. Harder, Chem. Rev., 2010, 110, 3852–3876 CrossRef CAS PubMed; (e) U. Kazmaier, Angew. Chem., Int. Ed., 2009, 48, 5790–5792 CrossRef CAS PubMed.
- (a) S. M. Li, I. Rashkov, J. L. Espartero, N. Manolova and M. Vert, Macromolecules, 1996, 29, 57–62 CrossRef CAS; (b) P. Dobrzyński, J. Kasperczyk and M. Bero, Macromolecules, 1999, 32, 4735–4737 CrossRef; (c) B. O'Keefe, J. M. A. Hillmyer and W. B. Tolman, Dalton Trans., 2001, 2215–2225 RSC; (d) Z. Zhong, P. J. Dijkstra, C. Birg, M. Westerhausen and J. Feijen, Macromolecules, 2001, 34, 3863–3868 CrossRef CAS; (e) M. H. Chisholm, J. Gallucci and K. Phomphrai, Chem. Commun., 2003, 48–49 RSC; (f) M. S. Hill and P. B. Hitchcock, Chem. Commun., 2003, 1758–1759 RSC; (g) M. Westerhausen, S. Schneiderbauer, A. N. Kneifel, Y. Söltl, P. Mayer, H. Nöth, Z. Zhong, P. J. Dijkstra and J. Feijen, Eur. J. Inorg. Chem., 2003, 3432–3439 CrossRef CAS PubMed; (h) M. H. Chisholm, J. C. Gallucci and K. Phomphrai, Inorg. Chem., 2004, 43, 6717–6725 CrossRef CAS PubMed.
- (a) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed; (b) Y. Sarazin, R. H. Howard, D. L. Hughes, S. M. Humphrey and M. Bochmann, Dalton Trans., 2006, 340–350 RSC; (c) D. J. Darensbourg, W. Choi, P. Ganguly and C. P. Richers, Macromolecules, 2006, 39, 4374–4379 CrossRef CAS; (d) D. J. Darensbourg, W. Choi and C. P. Richers, Macromolecules, 2007, 40, 3521–3523 CrossRef CAS; (e) M. G. Davidson, C. T. O'Hara, M. D. Jones, C. G. Keir, M. F. Mahon and G. Kociok-Kohn, Inorg. Chem., 2007, 46, 7686–7888 CrossRef CAS PubMed; (f) D. J. Darensbourg, W. Choi, O. Karroonnirun and N. Bhuvanesh, Macromolecules, 2008, 41, 3493–3502 CrossRef CAS; (g) C. A. Wheaton, P. G. Hayes and B. Ireland, Dalton Trans., 2009, 4832–4846 RSC; (h) V. Poirier, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Dalton Trans., 2009, 9820–9827 RSC; (i) Y. Sarazin, D. Rosca, V. Poirier, T. Roisnel, A. Silvestru, L. Maron and J.-F. Carpentier, Organometallics, 2010, 29, 6569–6577 CrossRef CAS; (j) C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 RSC; (k) D. Meimaroglou and C. Kiparissides, Macromolecules, 2010, 43, 5820–5832 CrossRef CAS; (l) X. Xu, Y. Chen, G. Zou, Z. Ma and G. Li, J. Organomet. Chem., 2010, 695, 1155–1162 CrossRef CAS PubMed; (m) Y. Sarazin, B. Liu, T. Roisnel, L. Maron and J.-F. Carpentier, J. Am. Chem. Soc., 2011, 133, 9069–9087 CrossRef CAS PubMed.
- (a) S. Harder, F. Feil and K. Knoll, Angew. Chem., Int. Ed., 2001, 40, 4261–4264 CrossRef CAS; (b) S. Harder and F. Feil, Organometallics, 2002, 21, 2268–2274 CrossRef CAS; (c) P. Jochmann, T. S. Dols, T. P. Spaniol, L. Perrin, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2009, 48, 5715–5719 CrossRef CAS PubMed.
- (a) A. G. M. Barrett, M. R. Crimmin, M. S. Hill and P. A. Procopiou, Proc. R. Soc. London, Ser. A, 2010, 466, 927–963 CrossRef CAS; (b) S. Harder, Chem. Rev., 2010, 110, 3852–3876 CrossRef CAS PubMed.
- (a) Y. M. Yamada and M. Shibasaki, Tetrahedron Lett., 1998, 39, 5561–5564 CrossRef CAS; (b) T. Suzuki, N. Yamagiwa, Y. Matsuo, S. Sakamato, K. Yamaguchi, M. Shibasaki and R. Noyori, Tetrahedron Lett., 2001, 42, 4669–4671 CrossRef CAS; (c) S. Saito and S. Kobayashi, J. Am. Chem. Soc., 2006, 128, 8704–8705 CrossRef CAS PubMed.
- (a) Y. M. A. Yamada and S. Ikegami, Tetrahedron Lett., 2000, 41, 2165–2169 CrossRef CAS; (b) A. Yamaguchi, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 10842–10843 CrossRef CAS PubMed; (c) Comprehensive asymmetric catalysis, ed. E. N.Jacobsen, A.Pfaltz and H.Yamamoto, Springer, Berlin, 1st edn, 1999 Search PubMed.
- (a) D. Almasi, D. A. Alonso and C. Nájera, Tetrahedron: Asymmetry, 2007, 18, 299–365 CrossRef CAS PubMed; (b) S. B. Tsogoeva, Eur. J. Org. Chem., 2007, 11, 1701–1716 CrossRef PubMed.
- (a) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829–2844 CrossRef CAS PubMed; (b) J. Christoffers and A. Baro, Angew. Chem., Int. Ed., 2003, 42, 1688–1690 CrossRef CAS PubMed; (c) O. M. Berner, L. Tedeschi and D. Enders, Eur. J. Org. Chem., 2002, 12, 1877–1894 CrossRef.
- (a) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3069 CrossRef CAS PubMed; (b) L. Susan, H. Jens and B. Armin, ChemCatChem, 2011, 3, 1708–1730 CrossRef PubMed.
- (a) S. Min and S. Wen-Sheng, Tetrahedron: Asymmetry, 1999, 10, 3319–3325 CrossRef; (b) S. Min and S. Wen-Sheng, Tetrahedron: Asymmetry, 2000, 11, 773–779 CrossRef; (c) S. Min and I. Yoshihisa, Aust. J. Chem., 2001, 54, 113–115 CrossRef; (d) K. K. Young, L. Tom and H. Yoshikazu, J. Am. Chem. Soc., 2003, 125, 9560–9561 CrossRef.
- (a) K. Naktode, R. K. Kottalanka and T. K. Panda, New J. Chem., 2012, 36, 2280–2285 RSC; (b) R. K. Kottalanka, K. Naktode and T. K. Panda, J. Mol. Struct., 2013, 1036, 188–195 CrossRef CAS PubMed; (c) R. K. Kottalanka, K. Naktode, S. Anga, H. P. Nayek and T. K. Panda, Dalton Trans., 2013, 42, 4947–4956 RSC; (d) R. K. Kottalanka, S. Anga, K. Naktode, P. Laskar, H. P. Nayek and T. K. Panda, Organometallics, 2013, 32, 4473–4482 CrossRef CAS; (e) R. K. Kottalanka, P. Laskar, K. Naktode, B. S. Mallik and T. K. Panda, J. Mol. Struct., 2013, 1047, 302–309 CrossRef CAS PubMed; (f) R. K. Kottalanka, A. Harinath, J. Bhattacharjee, H. V. Babu and T. K. Panda, Dalton Trans., 2014, 8757–8766 RSC; (g) J. Bhattacharjee, R. K. Kottalanka, A. Harinath and T. K. Panda, J. Chem. Sci., 2014, 126, 1463–1475 CrossRef CAS.
- (a) H. Brunner and R. G. Gastinger, J. Organomet. Chem., 1978, 145, 365–373 CrossRef CAS; (b) H. Brunner and G. O. Nelson, J. Organomet. Chem., 1979, 173, 389–395 CrossRef CAS; (c) E. Frauendorfer and H. Brunner, J. Organomet. Chem., 1982, 240, 371–379 CrossRef CAS.
- M. Wiecko, D. Girnt, M. Rastätter, T. K. Panda and P. W. Roesky, Dalton Trans., 2005, 2147–2150 RSC.
- T. K. Panda, M. T. Gamer and P. W. Roesky, Inorg. Chem., 2006, 45, 910–916 CrossRef CAS PubMed.
- The bonding situation in the drawing of the ligand system is simplified for clarity.
- T. K. Panda, H. Kaneko, O. Michel, H. Tsurugi, K. Pal, K. W. Törnroos, R. Anwander and K. Mashima, Organometallics, 2012, 31, 3178–3184 CrossRef CAS.
- C. Kling, H. Ott, G. Schwab and D. Stalke, Organometallics, 2008, 27, 5038–5042 CrossRef CAS.
- U. Englich and K. Ruhland-Senge, Z. Anorg. Allg. Chem., 2001, 627, 851–856 CrossRef CAS.
- H. Hao, S. Bhandari, Y. Ding, H. W. Roesky, J. Magull, H. G. Schmidt, M. Noltemeyer and C. Cui, Eur. J. Inorg. Chem., 2002, 1060–1065 CrossRef CAS.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Pergamon Press, Oxford, U.K., 1984 Search PubMed.
- (a) K. Ruhlandt-Senge, S. Davis, K. Dalal, U. Englich and M. O. Senge, Inorg. Chem., 1995, 34, 2587–2592 CrossRef CAS; (b) T. M. A. Al-Shboul, G. Volland, H. Görls, S. Krieck and M. Westerhausen, Inorg. Chem., 2012, 51, 7903–7912 CrossRef CAS PubMed.
- (a) J. A. Balanta-Díaz, M. Moya-Cabrera, V. Jancik, J. T. Morales-Juárez and R. Cea-Olivares, Polyhedron, 2013, 63, 167–172 CrossRef PubMed; (b) K. Ruhlandt-Senge and U. Englich, Chem.–Eur. J., 2000, 6, 4063–4070 CrossRef CAS.
- (a) B. J. Anderson, D. S. Glueck, A. G. DiPasquale and A. L. Rheingoldb, Organometallics, 2008, 27, 4992–5001 CrossRef CAS; (b) S. Hubert, A. Stützer, P. Bissinger and A. Schier, Z. Anorg. Allg. Chem., 1993, 619, 1519–1525 CrossRef PubMed.
- T. P. Hanusa, in Comprehensive Organometallic Chemistry III, ed. R. H.Crabtree and M. P.Mingos, Elsevier, Oxford, 2007, vol. 2, p. 67 Search PubMed.
- (a) T. P. Hanusa, Organometallics, 2002, 21, 2559–2571 CrossRef CAS; (b) T. P. Hanusa, Chem. Rev., 2000, 100, 1023–1036 Search PubMed; (c) T. P. Hanusa, Coord. Chem. Rev., 2000, 210, 329–367 CrossRef CAS.
- T. K. Panda, K. Yamamoto, K. Yamamoto, H. Kaneko, Y. Yang, H. Tsurugi and K. Mashima, Organometallics, 2012, 31, 2268–2274 CrossRef CAS.
- M. Kuzdrowska, L. Annunziata, S. Marks, M. Schmid, C. G. Jaffredo, P. W. Roesky, S. M. Guillaume and L. Maron, Dalton Trans., 2013, 42, 9352–9360 RSC.
- B. Liu, T. Roisnel, J.-P. Guegan, J.-F. Carpentier and Y. Sarazin, Chem.–Eur. J., 2012, 18, 6289–6301 CrossRef CAS PubMed.
- Y. Sarazin, B. Liu, L. Maron and J.-F. Carpentier, J. Am. Chem. Soc., 2011, 133, 9069–9087 CrossRef CAS PubMed.
- M. G. Davidson, C. T. O'Hara, M. D. Jones, C. G. Keir, M. F. Mahon and G. Kociok-Köhn, Inorg. Chem., 2007, 46, 7686–7688 CrossRef CAS PubMed.
- (a) I. Palard, A. Soum and S. M. Guillaume, Chem.–Eur. J., 2004, 10, 4054–4062 CrossRef CAS PubMed; (b) J. Jenter, P. W. Roesky, N. Ajellal, S. M. Guillaume, N. Susperregui and L. Maron, Chem.–Eur. J., 2010, 16, 4629–4638 CrossRef CAS PubMed.
- M. Sheldrick, SHELXS-97, Program of Crystal Structure Solution, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program of Crystal Structure Refinement, University of Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1053400–1053411. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra04495b |
| This journal is © The Royal Society of Chemistry 2015 |