
Study of the Diels–Alder and retro-Diels–Alder reaction between furan derivatives and maleimide for the creation of new materials†
Abstract
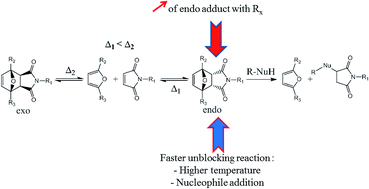
The Diels–Alder reaction leads to a mixture of two diastereomers, one called endo and the other one exo. The cyclo-reversion temperature of the first one is lower than the exo adduct and the ratio between endo and exo adducts varies according to the substituents of the Diels–Alder partners and experimental parameters. Therefore, the influence of some reaction parameters such as the substituents of furan and maleimide derivatives, the reaction temperature and the presence of a nucleophile on the endo/exo Diels–Alder ratio and/or the retro-Diels–Alder reaction have been studied. For instance, furan and maleimide derivatives with electron withdrawing substituents induced the creation of the endo adduct preferentially. Also the presence of a far electron withdrawing substituent on furan and/or an electron attracting mesomeric substituent on maleimide resulted in a faster reversibility of the endo adduct. Finally, a high temperature and the presence of a nucleophile (thiol) also induced faster retro-Diels–Alder kinetics. Moreover, it was proved that isomerization from the endo to the exo diastereomer is preceded by a retro-Diels–Alder reaction of the endo adduct. The presence of a nucleophile in the mixture confirmed this result. This study allowed the highlighting of different parameters of the Diels–Alder reaction to obtain as much endo adduct as possible, and a fast and/or full retro-Diels–Alder reaction of this adduct.
 Please wait while we load your content...
Please wait while we load your content...

