
Supramolecular hydrogels of α-cyclodextrin/reverse poloxamines/carbon-based nanomaterials and its multi-functional application†
Abstract
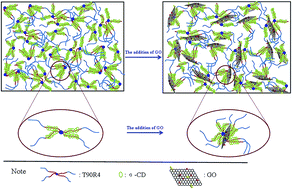
Supramolecular hydrogels were prepared using α-cyclodextrin (α-CD) and a poloxamine (reverse Tetronic 90R4, T90R4) which has four diblock arms with a poly(propylene oxide)–poly(ethylene oxide) (PPO–PEO) structure. The α-CD can slide past the PPO blocks and towards the middle PEO blocks owing to the unsuitable energy between α-CD and PPO to form α-CD/T90R4 inclusion complexes (ICs). The incorporation of graphene oxide (GO) into α-CD/T90R4 ICs changes their phase behavior and forms mechanically strong hydrogels because of the hydrogen-bonding between the GO nanosheets and the α-CD and PEO blocks of T90R4. The native hydrogel, as well as the α-CD/T90R4/GO hybrid hydrogels, have been thoroughly characterized by using various microscopy techniques. Field emission scanning electron microscopy (FE-SEM) was used to observe the morphology of the hydrogel, and differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) were used to evaluate the thermal stability of the hydrogel. Fourier transform infrared (FT-IR) spectroscopy and X-ray diffraction (XRD) were used to characterize the interactions within the supramolecular assemblies and the degree of crystallinity of the xerogels, respectively. The experimental results demonstrated that α-CD/T90R4/GO hybrid hydrogels could adsorb various dyes selectively, and that it is a promising candidate for sewage treatment, while the native one cannot adsorb the dyes well. Moreover, the native α-CD/T90R4 hydrogel has excellent biocompatibility, and the results of the in vitro drug release study showed that the injectable doxorubicin (DOX)-loaded hydrogel is appropriate for the controlled release of anticancer drugs, while the α-CD/T90R4/GO hybrid hydrogels can reduce the release rate of DOX.
 Please wait while we load your content...
Please wait while we load your content...

