
Integrated electrospun carbon nanofibers with vanadium and single-walled carbon nanotubes through covalent bonds for high-performance supercapacitors†
Kexin Tangabc,
Yuping Li*b,
Hongbin Caoabc,
Feng Duanb,
Jason Zhanga,
Yi Zhangabc and
Yi Wangb
aNational Engineering Research Centre for Distillation Technology, School of Chemical Engineering and Technology, Tianjin University, Tianjin 300072, PR China
bKey Laboratory of Green Process and Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, PR China. E-mail: ypli@ipe.ac.cn; Fax: +86-10-82544844-816; Tel: +86-10-82544844-810
cCollaborative Innovation Centre of Chemical Science and Engineering, Tianjin University, Tianjin 300072, PR China
First published on 21st April 2015
Abstract
We report a ternary composite vanadium/single-walled carbon nanotubes (SWCNTs)/carbon nanofibers (VSCNFs) material using hybrid-electrospinning and carbonizing of polyacrylonitrile, polyvinylpyrrolidone, SWCNTs and vanadyl acetylacetonate. The morphology and structure of the ternary composites are characterized. Its electrochemical properties are measured in a 6 mol L−1 aqueous KOH electrolyte. VSCNFs possesses a hierarchical structure with micropores and mesopores and a specific surface area of 821 m2 g−1, and it also exhibits a reversible specific capacitance of 479 F g−1 at 1 A g−1 and 367.4 F g−1 at 10 A g−1, and retains 94% of its initial capacitance after 5000 cycles (8 A g−1). The results show that simultaneously adding vanadium and SWCNTs can greatly enhance the conductivity, capacitive performance and stability by forming a closer connection with nanofibers through V–N–C, V–O–C and V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds. SWNCTs are not only mechanically mixed in CNFs to enhance the extent of graphitization, but also involved in the transmutation process of CNFs graphitization with the participation of vanadium.
O bonds. SWNCTs are not only mechanically mixed in CNFs to enhance the extent of graphitization, but also involved in the transmutation process of CNFs graphitization with the participation of vanadium.
1. Introduction
To meet the increasing demands for clean and sustainable energy, high-capacity energy storage devices like supercapacitors have attracted much attention.1 According to the charge storage mechanisms, three types of supercapacitors can be summarized:2 electronic double layer (EDL) capacitors, pseudo-capacitors and hybrid capacitors. An EDL capacitor usually possesses a short charge time, long cycling life and high power density, but its storage capabilities need to be improved. In comparison, a pseudo-capacitor has a higher current response due to the chemical reactions on the interface of electrode, while the disappointing capacitance retention limits its further application. Consequently, a hybrid capacitor, that combines both the advantages of an EDL capacitor and pseudo-capacitor, has been developed to obtain extraordinary performances.Due to the abundant storage, low cost and relative high structure stability, carbon materials are most commonly used in supercapacitors,3,4 such as activated carbon,5 carbon nanofibers,6 carbon aerogels,7 ordered mesoporous carbon,8 carbon nanotubes,9 carbon black10 and graphene,11 especially two or more carbon materials are composited together to achieve sophisticated structure and characteristics,4,12,13 like three-dimensional network,14 where activated carbon (AC), graphene or carbon nanofibers (CNFs) usually acts as carbon skeleton, and carbon nanotubes (CNTs) or carbon black (CB) acts as ancillary elements. For example, single-walled carbon nanotubes (SWCNTs) are sometimes used as additive for CNFs electrode because of its high conductivity and large effective specific surface area,15 and it also can help to build hierarchical pore structure.16
Metal oxides such as TiO2, RuO2, MnO2, and VOx, are widely used to modify carbon materials to get pseudo-capacitance.17,18 RuO2 is reported to be one of the most effective additives, but its high price limited its application.5,19 TiO2 composite carbon nanofibers are more likely used in photocatalysis research.20,21 One of the most development MnO2 modified carbon electrode show relative low capacitance.22 V2O5 (vanadium pentoxide),23–25 VO2 (vanadium dioxide)26,27 and organic vanadium28 are reported to modify porous carbon electrode, while the insulation of these compound weaken carbon nanofibers conductivity and pore structure, but still vanadium compounds might be a suitable additives due to its relatively low cost and abundance in China.29 There are two distinctive formations of vanadium compound inserted in the carbon electrode, namely, amorphous vanadium and crystal vanadium oxide. Some studies show that amorphous vanadium may exhibit more superior capacitive properties compared to crystal vanadium,27,28,30 because amorphous vanadium is likely linked to carbon skeleton with covalent bonds in atom level, and thus enhance the contact of transition metal with ions. In comparison, the crystal vanadium is physically dispersed in the carbon skeleton. Therefore, the research on single-step synthesis and simultaneously incorporating vanadium and conductive additives to form an integrated structure with tunable pore distribution and efficient surface area and good conductivity may be a possible approach to achieve high performance and steady supercapacitor.
Electrospinning technique is a simple and versatile method for producing continuous nanofibers from polymers, composites or ceramics.31 In this work, we have successfully synthesized a composite vanadium/SWCNTs/CNFs electrode for supercapacitors with hybrid electrospinning and carbonization techniques. The fabricated composite CNFs (VSCNFs) have achieved advantages as follows: (1) hierarchical structure, which can enhance ion diffusion within CNFs; (2) unique V–N–C bond to combine carbon skeleton and graphite fragments; (3) high extent of graphitization to improve structure regularity and conductivity; (4) high capacitance, and stable electrochemical cyclic properties. This work provides a performable method of synthesizing stably composited metal–carbon materials and predicts the vanadium and SWCNTs involved structure.
2. Experimental
2.1 Materials preparation
Polyacrylonitrile (PAN, Mw = 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was purchased from Sigma-Aldrich (USA). Polyvinylpyrrolidone (PVP, Mw = 1300000) was obtained from Aladdin (China). Vanadyl (IV) acetylacetonate (VO(acc)2, purity > 98%) was bought from J&K (China). All other chemicals used in this work were purchased from Beijing Chemical Industry Group (China). Single-walled carbon nanotubes (SWCNTs, purity > 95%, OD < 2 nm) were purchased from Shenzhen Nanotech Port (China). SWCNTs were simply treated with diluted nitric acid to remove little residential metal catalyst.
000) was purchased from Sigma-Aldrich (USA). Polyvinylpyrrolidone (PVP, Mw = 1300000) was obtained from Aladdin (China). Vanadyl (IV) acetylacetonate (VO(acc)2, purity > 98%) was bought from J&K (China). All other chemicals used in this work were purchased from Beijing Chemical Industry Group (China). Single-walled carbon nanotubes (SWCNTs, purity > 95%, OD < 2 nm) were purchased from Shenzhen Nanotech Port (China). SWCNTs were simply treated with diluted nitric acid to remove little residential metal catalyst.
| Samples | Pure PAN/PVP | SWCNTs + PAN/PVP | VO(acc)2 + PAN/PVP | VO(acc)2 + SWCNTs + PAN/PVP | ||
|---|---|---|---|---|---|---|
| 10% VO(acc)2 | 15% VO(acc)2 | 20% VO(acc)2 | ||||
| Precursor nanofibers | PNFs | SNFs | VNFs | 10VSNFs | 15VSNFs | 20VSNFs |
| Carbonized at 800 °C | PCNFs-800 | SCNFs-800 | VCNFs-800 | 10VSCNFs-800 | 15VSCNFs-800 | 20VSCNFs-800 |
| Carbonized at 900 °C | PCNFs-900 | SCNFs-900 | VCNFs-900 | 10VSCNFs-900 | 15VSCNFs-900 | 20VSCNFs-900 |
2.2 Morphology and structure characterization
The morphology and element contents of samples were observed by a field emission scanning electron microscopy (FE-SEM, JSM-7001F, JEOL, Japan) equipped with energy dispersive spectrometer (EDS, INCA X-MAX, Oxford Instruments, UK). Thermal gravity analysis and differential scanning calorimetry was performed in a TGA/DSC1 STARe System (Metler Toledo, Switzerland). High resolution field emission transmission electron microscopy (TEM, JEM 2100F, JEOL, Japan) was employed to observe samples inner structure. X-ray diffraction (XRD) was conducted on SmartLab (Rigaku, Japan). Raman spectroscopy was obtained on LabRAM HR800 (Horiba Jobin Yvon, French). Fourier transform infrared spectroscopy (FT-IR) was obtained on a Spectra spectrometer (Nicolet iS50, Thermal Fisher Scientific, USA). X-ray photoelectron spectroscopy (XPS) was measured using ESCALAB 250Xi spectrometer (Thermo Fisher Scientific, United States). Nitrogen sorption isotherm curves of samples were obtained by automatic specific surface analyzer (Quantachrome, United States) at liquid nitrogen boiling point of 77 K, and the samples were outgassed at 200 °C for 8 h before measurement. Brunauer–Emmett–Teller (BET) method was used to calculate the specific surface area (SBET). Pore size distribution (PSD) and total pore volume was calculated by the density function theory (DFT), and the volume of micropores was provided by Horvath–Kawazoe (HK) method.2.3 Electrochemical measurements
Samples electrochemical performances were studied in a typical three-electrode system on Autolab electrochemical workstation (PGSTAT302N, Metrohm, Switzerland). The working electrode (WE) was fabricated by pressing activated materials and nickel foams under a pressure of 10 MPa for 1 min, where the as-synthesized materials act as active composite and nickel foams act as current collector, and the size of WE was 1 cm × 1 cm × 260 μm (contains two layers of pressed nickel foams and one layer of activated materials), and the average mass for activated materials is about 3 mg. The reference electrode was Hg/HgO electrode immersed in 1 M potassium hydroxide aqueous solution (KOH). The counter electrode (CE) was platinum sheet with an area of 1 cm2. The electrolyte was 6 M KOH. Cyclic voltammetry (CV) measurements were performed in a voltage window from −1.0 to 0 V (vs. Hg/HgO) at scan rates of 5, 10, 20, 30, 40, 50, 80 to 100 mV s−1. Galvanostatic charge–discharge (GCD) tests were conducted at current densities of 1, 2, 5, and 10 A g−1 based on active materials weight of WE. Electrochemical impedance spectroscopy (EIS) information was obtained in range of 105–0.01 Hz at open circuit potentials with an amplitude signal model of 5 mV.The specific capacitance (F g−1) obtained from CV method is based on eqn (1):
 | (1) |
The specific capacitance (F g−1) derived from GCD curves (mean value over discharging time) is calculated from the eqn (2):
 | (2) |
3. Results and discussion
3.1 Morphology and structure
Uniform diameter distribution and continuous nanofibers of precursor and carbonized samples are observed from Fig. S1 (ESI†), indicating stable electrospinning and carbonizing process. The FE-SEM images show the smooth surface of PNFs and SNFs without apparent black spot or agglomeration (Fig. 1a1 and b1), indicating that the activation and oxidation of SWCNTs are helpful for regular distribution in the nanofibers. Contrastively, VNFs and VSNFs present rougher surface, and the surface become smooth after carbonization (Fig. 1c and d). The average diameter of SNFs (540 nm) is smaller than others (Table S2†), suggesting additives conductivity influences precursor nanofibers diameter, because the conductivity of SWCNTs is better than the organic compound VO(acc)2.34–37 After pre-oxidation and carbonization, CNFs are dramatically shrunk and overlapped. Notably, the CNFs overlapping would greatly enhance the conductivity of CNFs webs. Table 2 presents the elementary composition of composite nanofibers. The carbon ratio of the samples is increased after adding SWCNTs and decreased after loading VO(acc)2, and 15VSCNFs-800 has nearly the same nitrogen and oxygen content with VCNFs-800, only except substituting amount of 4 wt% vanadium for carbon. Carbonization degree is enhanced by increasing annealed temperature. However, the excessive cyclization and cross-linking reactions of polymers in higher temperature may cause surface defects (Fig. 1d3). The vanadium load in 15VSCNFs-800 is 8%, and its atom ratio of carbon, oxygen, nitrogen and vanadium is 39:5:3:1. Notably, nitrogen element content is significantly decreased after carbonization, but it's still a significant content that can influence electrode performance25,38 due to the surplus electron pair of nitrogen. Consequently, uniform composite CNFs webs have been obtained through electrospinning and carbonization, and SWCNTs and vanadium have been successfully inserted into nanofibers.
 | ||
| Fig. 1 Field emission scanning electron microscopy images of nanofibers: (a1–a3) PNFs; PCNFs-800 and PCNFs-900; (b1–b3) SNFs, SCNFs-800 and SCNFs-900; (c1–c3) VNFs, VCNFs-800 and VCNFs-900; (d) 15VSNFs, 15VSCNFs-800 and 15VSCNFs-900. | ||
| Atom | PNFs | PCNFs | SNFs | SCNFs | VNFs | VCNFs | 15VSNFs | 15VSCNFs | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 800 | 900 | 800 | 900 | 800 | 900 | 800 | 900 | |||||
| C | 67.7 | 81.8 | 86.8 | 67.0 | 85.0 | 90.2 | 69.2 | 78.0 | 81.6 | 67.0 | 74.0 | 82.5 |
| N | 22.5 | 12.1 | 5.8 | 22.9 | 7.4 | 3.6 | 18.1 | 6.5 | 4.2 | 19.9 | 6.3 | 4.8 |
| O | 9.8 | 6.1 | 7.4 | 10.1 | 7.6 | 6.2 | 11.6 | 10.2 | 8.8 | 11.4 | 11.7 | 4.2 |
| V | 0 | 0 | 0 | 0 | 0 | 0 | 1.1 | 5.3 | 5.4 | 1.7 | 8.0 | 8.5 |
Fig. 2 displays the N2 sorption isotherm curves of as-synthesized samples (800 °C). PCNFs electrode exhibits typical type-I Langmuir adsorption behaviour (Fig. 2a) according to IUPAC, and other five samples possess hierarchical structure with micropores and mesopores (Fig. 2b), which can be distinguished through the small adsorption hysteresis during pressure P/P0 from 0.8 to 0.3 as well as the strong adsorption behaviour during low pressure (P/P0 < 0.1). As shown in Table 3, SCNFs (696 m2 g−1) have slightly larger specific surface area (SBET) compared to PCNFs (642 m2 g−1), and the SBET of VCNFs is slightly increased after inserting vanadium, but the micropores volume ratio is decreased, which impairs electrochemical performance,2 and adding vanadium and SWCNTs into PCNFs can hold SBET and micropores volume ratio simultaneously. This can be explained as follows: according to ref. 28, 32 and 39, carbonization process of composite PAN/PVP nanofibers contains two pathways: (I) polymer cross-linking and pyrolysis; (II) competitive reactions between VO(acc)2 and polymer skeleton. Carbonization process of PNFs and SNFs without VO(acc)2 is driven by pathway I, resulting almost pure micropores, meanwhile, VNFs and VSNFs are driven by both pathway I and II, resulting expanded mesopores. On the other hand, the opened SWCNTs with inner diameter less than 2 nm can increase the ratio of micropores and the specific surface area, and the external structure of SWCNTs may also facilitate the generation of mesopores to enlarge the ion channel. However, excessive VO(acc)2 (20 wt%) overly strengthen pathway II, resulting serious SBET and micropores volume ratio decline.
 | ||
| Fig. 2 Nitrogen isothermal adsorption analysis: (a) nitrogen adsorption isotherm curves and (b) pore size distribution plots of samples carbonized at 800 °C. | ||
| Samples | PCNFs | SCNFs | VCNFs | 10VSCNFs | 15VSCNFs | 20VSCNFs | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Temp. | 800 | 900 | 800 | 900 | 800 | 900 | 800 | 900 | 800 | 900 | 800 | 900 |
| a SBET is abbreviation of the specific surface area calculated from Brunauer–Emmett–Teller (BET) method, and Vmic and Vtotal are the effective adsorption volume of micropores and total volume. | ||||||||||||
| SBET (m2 g−1) | 642 | 683 | 696 | 730 | 721 | 106 | 817 | 343 | 821 | 168 | 418 | 101 |
| Vmic (cm3 g−1) | 0.292 | 0.302 | 0.280 | 0.292 | 0.184 | 0.052 | 0.285 | 0.082 | 0.310 | 0.063 | 0.178 | 0.041 |
| Vtotal (cm3 g−1) | 0.321 | 0.327 | 0.370 | 0.388 | 0.308 | 0.194 | 0.395 | 0.269 | 0.470 | 0.218 | 0.327 | 0.183 |
| Vmic/Vtotal (%) | 91.0% | 92.4% | 75.7% | 75.3% | 59.7% | 26.8% | 72.2% | 30.5% | 66.0% | 28.9% | 54.4% | 22.4% |
Carbonizing temperature could affect the pore structure of CNFs as well. When raising carbonizing temperature from 800 °C to 900 °C, the SBET of PCNFs and SCNFs increase from 642, 696 to 683, 730 m2 g−1, respectively. However, the SBET of VCNFs, VSCNFs decrease dramatically. This may due to the enhanced reaction of pathway II when raising temperature. These results indicate that raising carbonized temperature is a feasible method to enhance structure property of vanadium-free carbon materials, but to vanadium-composited sample, carbonized temperature varying method is undesirable.
The transmission electron microscopy (TEM) images show the morphology and surface pore structure of PCNFs-800, SCNFs-800 and 15VSCNFs-800 (Fig. 3 and S9†). PCNFs-800 and SCNFs-800 present denser structure than 15VSCNFs-800 although they have similar diameter around 350 nm. The reason is that 15VSCNFs-800 possesses larger ratio of mesopores than PCNFs and SCNFs (Fig. 2b), and it is supposed that the mesopores in 15VSCNFs-800 lead some electron beams generated by laser in TEM to penetrate the edge of CNFs and finally to present looser internal structure. The core of CNFs shows black possibly because those mesopores are arranged irregular and vermicular40,41 (Fig. 3d and S9d†).
 | ||
| Fig. 3 Transmission electron microscopy images: (a) PCNFs-800, (b) SCNFs-800, (c) 15VSCNFs-800 and (d) high magnification image of 15VSCNFs-800. | ||
Fig. 4 shows the Raman spectroscopy of the composited CNFs. Two peaks at 1360 cm−1 and 1580 cm−1 are obtained (Fig. 4a), corresponding to the D-band for amorphous carbon and G-band for orderly graphitized carbon, respectively.43,44 The ratio of relative intensity of D-band and G-band (ID/IG), which represents the extent of carbon defects, is summarized in Fig. 4b. For SWCNT-free CNFs, the ID/IG value of VCNFs-800 is much higher than that of PCNFs. On the contrary, no obvious increase of ID/IG is observed after embedding SWCNTs. ID/IG of samples at 900 °C is smaller than that at 800 °C except for 15VSCNFs (Fig. 4b), suggesting that the graphitic sheets should be continuously growing when carbonized temperature is increased, and for 15VSCNFs, the exothermic reactions between VO(acc)2 and PAN/PVP may be accelerated when carbonized temperature and VO(acc)2 content were increased simultaneously. Therefore, adding 15% VO(acc)2 and 10% SWCNTs within electrospun precursors is a highly effective approach to improve the graphitizing extent of composite CNFs.
 | ||
| Fig. 4 Analysis of carbon defects of samples: (a) Raman spectra and (b) the ratio of ID and IG (ID/IG) of PCNFs, SCNFs, VCNFs and 15VSCNFs (the solid line and short dot line for (a) represent samples carbonized at 800 °C or 900 °C, respectively). | ||
The effects of SWCNTs content, VO(acc)2 content and carbonizing temperature on crystal structure of CNFs are studied by XRD (Fig. 5). Two diffractive peaks are observed at 2θ ≈ 25° and 43°, which are attributed to graphitic structure (0 0 2) and turbostratic carbon structure (1 0 1), respectively.44 It's obvious that the (0 0 2) peak gets sharper and gradually shifts right (2θ ≈ 26°) when SWCNTs are added into CNFs, confirming that SWCNTs have been successfully loaded. The (0 0 2) peak of CNFs also turns sharper and stronger after loading vanadium; this can be explained that vanadium may participate graphitizing process and enhance the extent of graphitization. Furthermore, vanadium in CNFs may be amorphous as vanadium oxide peaks are failed to be detected.30 The (0 0 2) peak and (1 0 1) peak are both slightly magnified when carbonizing temperature is increased from 800 °C to 900 °C, confirming the graphitization and turbostratic structure are both enhanced. The Raman and XRD results show that SWNCTs are not only mechanically mixed in CNFs to enhance the extent of graphitization, but also involved in the transmutation process of CNFs graphitization with the participation of VO(acc)2.
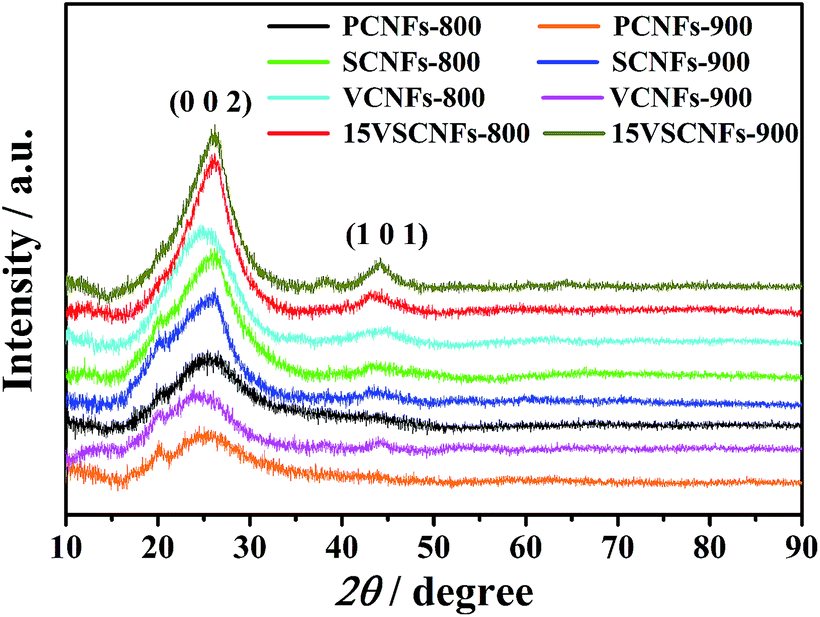 | ||
| Fig. 5 X-ray diffraction curves of PNFs, SCNFs, VCNFs, and 15VSCNFs carbonized at 800 °C or 900 °C. The (0 0 2) peak and (1 0 1) peak represents graphitic structure and turbostratic carbon structure, respectively. | ||
3.2 Integrated structure with vanadium and SWCNTs involved
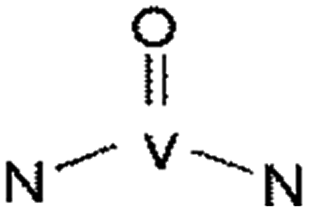
The morphological and structure characterizations suggest that there is an integrated structure formed between vanadium and carbon skeleton with participating of SWCNTs. For clearly understanding of the structure and chemical bond between vanadium and carbon skeleton, XPS spectrum of 15VSCNFs-800 is obtained (Fig. 6). The peaks of carbon (C1s), oxygen (O1s), nitrogen (N1s) and vanadium (V2p) are observed at the survey region (Fig. 6a). The C1s spectrum shows four peaks at 284.7 eV, 285.4 eV, 286.7 eV and 289.7 eV, corresponding to C–C, C–N, C–O, and CO bonds,45 respectively (Fig. 6b). Nitrogen may exist in four types, which are N–V (398.9 eV), R–N (401.5 eV), C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N and EWGs–N (electron withdrawing groups) (402.2 eV)46 (Fig. 6c). For the vanadium element, it could link to carbon skeleton through V–O–C and VO bonds.28 However, V–N–C bond may also exist in CNFs. The binding energy of V2p3/2, N1s, and C1s is consistent with literature42 (Fig. 6e), and Table 4 illustrates the corresponding binding energy; this structure could strengthen CNFs by offsetting the defects caused by the pyrolysis of PAN/PVP and VO(acc)2. Notably, the V–C bond is unlikely to exist as no peaks are observed at 282.5 eV for C1s and 513.5 eV for V2p3/2.47
N and EWGs–N (electron withdrawing groups) (402.2 eV)46 (Fig. 6c). For the vanadium element, it could link to carbon skeleton through V–O–C and VO bonds.28 However, V–N–C bond may also exist in CNFs. The binding energy of V2p3/2, N1s, and C1s is consistent with literature42 (Fig. 6e), and Table 4 illustrates the corresponding binding energy; this structure could strengthen CNFs by offsetting the defects caused by the pyrolysis of PAN/PVP and VO(acc)2. Notably, the V–C bond is unlikely to exist as no peaks are observed at 282.5 eV for C1s and 513.5 eV for V2p3/2.47
 | ||
| Fig. 6 X-ray photoelectron spectroscopy of 15VSCNFs-800: (a) survey scan, and peak curves of (b) C1s, (c) N1s, (d) O1s, and (e) V2p. | ||
| Element | N1s | O1s | V2p3/2 | V2p1/2 | |
|---|---|---|---|---|---|
 |
Ferrer42 | 399.1 | 532.9 | 516.5 | 524.3 |
| This work | 398.9 | 532.9 | 516.4 | 524.4 | |
TGA/DSC method is conducted to promote the understanding of the thermal behaviours after inserting vanadium and SWCNTs. As shown in Fig. 7, the sharp exothermic peaks around 300 °C are observed obviously, mainly due to the cyclization and oxidation reaction of PAN and PVP,32 and the small endothermic peak around 480 °C is caused by dehydrogenation and excessive oxidation of PAN, and the wide exothermic peak from 500 °C to 800 °C is about the further dehydrogenation and intensively cyclization of carbon skeleton.32 The decomposition of PVP mainly happens at 600 °C.39 With the increase of vanadium content, the weight loss rate slows down, and SWCNTs barely influence the weight loss (Fig. 7a). The embedded vanadium could influence carbonizing process in two competitive ways: firstly, VO(acc)2 would be connected to carbon skeleton during the pyrolysis process of PAN and PVP, which may produce micropores and mesopores with emitting of N2, CO, CO2, HCN, etc.,32 resulting in accelerating the pyrolysis process (Fig. 7b) and more serious weight loss. Secondly, the replacement of C, N, H atoms with vanadium atom which has higher relative molecular weight would greatly slow down the weight loss. Fig. 7c displays the Raman spectra of 15VSCNFs-800. Four distinguishable variations are observed for the following Raman peaks: 1010 [v(VO)], 749 [vas(V–O)], 501 [vs(V–O)], and 267 cm−1 [v(V–N)], which are in accordance with ref. 48 and 49. Combining with analysis from XPS, this provides the direct information about the new formed V–O–C, V–N–C and VO bonds.
 | ||
| Fig. 7 Thermal behaviour analysis of electrospun precursors: (a) thermal gravity analysis and (b) differential scanning calorimetry curve of precursor nanofibers: PNFs, VNFs, 10VSNFs and 20VSNFs with N2 following at 60 mL min−1 with heating rate of 10 °C min−1. (c) Raman spectra of 15VSCNFs-800 with wavenumber from 1100 to 150 cm−1. (d) Predicted integrated graphite structure of V–N–C and V–O–C, and VO bonds involved in carbon skeleton. | ||
Based on above discussion, we predict the vanadium-involved graphite skeleton structure (Fig. 7d). According to the atom ratio of 15VSCNFs-800, there are three kinds of possible structure: V–O–C and V–N–C can link graphene fragments into large graphite sheets, or V4+ substitute for carbon atom, or V3+ bond with carbon skeleton margin through (O)′V–O–C form to cause faradic response when applying for capacitive devices. Notably, V–O–C and V–N–C covalent bonds may not only exist between fragments, but also bond between two layers to form multiple layers graphite. This structure can store more electrons when charging the electrode. However, due to the molecular structure of carbon, nitrogen and vanadium, the desultory carbon skeleton would wave naturally; consequently this structure would form much steric hindrance for storing electrons and lead to increase of carbon disorder and amorphous, so SWCNTs (OD < 2 nm) can support between carbon skeleton to separate layer, prompt graphite flake growth and act as conductive tunnel.
3.3 Electrochemical performance
Cyclic voltammetry (CV) method is a direct way to characterize electrodes capacitive ability. Fig. 8 describes CV curves of PCNFs-800, SCNFs-800, VCNFs-800 and 15VSCNFs-800. The CV shape of SCNFs-800 is closer to rectangle than that of PCNFs-800 (Fig. 8a and b), and the as-calculated CCV of SCNFs-800 is improved with increasing scan rates, however, CCV rate performance of PCNFs-800 is reduced, indicating that the former is closer to the ideal double layer supercapacitor behaviour.44 The CV curves of VCNFs-800 and 15VSCNFs-800 are deformed seriously as compared to PCNFs-800 (Fig. 8c and d), indicating that adding vanadium without SWCNTs reduces the stability of electrode when increasing scan rates, but SWCNTs can make up capacitive performance by enhancing current response due to the developed porous integrated structure. | ||
| Fig. 8 Cyclic voltammetry curves: (a) PCNFs-800, (b) SCNFs-800, (c) VCNFs-800 and (d) 15VSCNFs-800 within scan rates from 5 to 100 mV s−1, (e) CV scanning at 5 mV s−1, (f) CCV as a function of scan rates. | ||
The two peaks of VCNFs-800 and 15VSCNFs-800 at reduction potential of −0.9 V and oxidation potential of −0.3 V are possibly corresponding to the redox of vanadium-related groups in composite CNFs,28 suggesting pseudo-capacitive behaviour (Fig. 8e). The CV area at 5 mV s−1 is increased from PCNFs, SCNFs, VCNFs to 15VSCNFs, suggesting both SWCNTs and vanadium can enhance PCFNs capacitive performance, but in different ways: the former enhance conductivity, and the latter introduce pseudo-capacitance. As predicted previously, the integrated 15VSCNFs combine these two key factors, in result of even larger CV area. The capacitive rate performance of 15VSCNFs-800 is not as good as PCNFs and SCNFs, probably due to pseudo-capacitive behaviour with 8% vanadium. However, it has CCV specific capacitance of 264 F g−1 at scan rate of 5 mV s−1, which is much higher than other samples. These results indicate the positive effects of incorporating vanadium and SWCNTs into CNFs on enhancing the conductivity and capacitive performance of CNFs.
The GCD curves of PCNFs, SCNFs, VCNFs and 15VSCNFs at varying current density from 1 to 10 A g−1 are demonstrated in Fig. 9, where the charge–discharge cycling period has been significantly increased by compositing SWCNTs and vanadium in CNFs, and the corresponding GCD curves for CNFs-800 at a constant current density of 2 A g−1 are also obtained (Fig. 9e). PCNFs and SCNFs have almost the same GCD curves, but still the discharging time of SCNFs-800 (206 F g−1) is slightly increased, which is 10.2% higher than that of PCNFs-800 (187 F g−1). Notably, both VCNFs and 15VSCNFs have a turning point at −0.75 V for discharging and another at −0.38 V for charging, corresponding to redox reactions caused by the embedded vanadium(IV), and the CGCD of 15VSCNFs-800 (428 F g−1 at current density of 2 A g−1) is higher than that of VCNFs-800 (305 F g−1 at current density of 2 A g−1) because of the integrated structure formed by inserting vanadium and SWNCTs into CNFs.
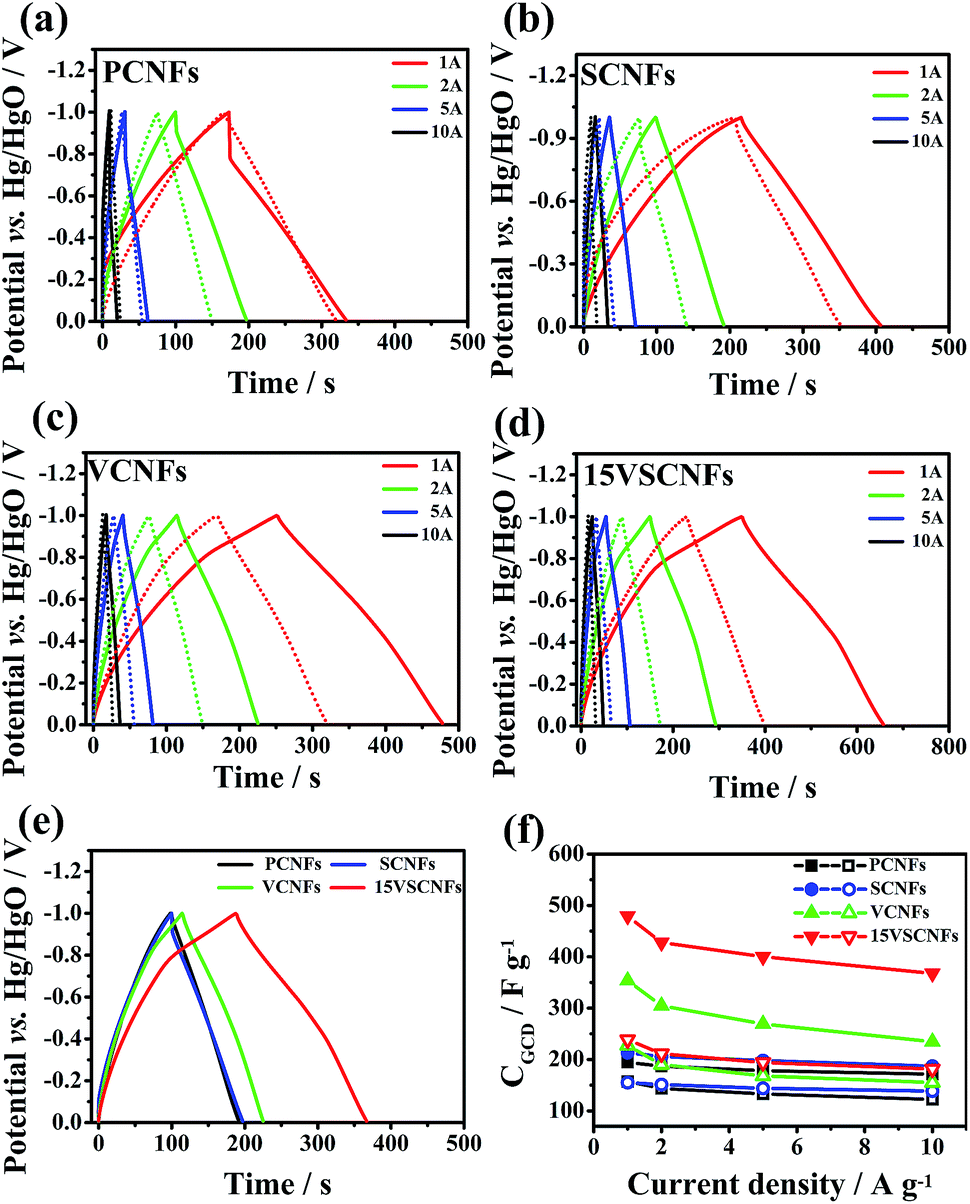 | ||
| Fig. 9 Galvanostatic charge–discharge (GCD) curves: (a) PCNFs, (b) SCNFs, (c) VCNFs and (d) 15VSCNFs with current densities from 1 to 10 A g−1, and (e) the corresponding GCD curves at 2 A g−1 carbonized at 800 °C, and (f) the specific capacitance (CGCD) plots as function of current density (the solid line and the short dash line for (a) to (d) represents at 800 °C and 900 °C, respectively). | ||
A good supercapacitor should be able to perfectly work at high current density. Fig. 9f shows the specific capacitance retention behaviour. 15VSCNFs-800 exhibits the highest specific capacitance of 479 F g−1 at current density of 1 A g−1 and 367.4 F g−1 at current density of 10 A g−1, where 76.7% of initial capacitance at 1 A g−1 is remained at high density of 10 A g−1, and samples of PCNFs-800 and SCNFs-800 keep 88.1% and 87.8% of initial capacitance, respectively.
As shown in Table S3,† in terms of capacitance rate performance, VCNFs electrode is worse than 15VSCNFs. With the same vanadium loading, 10VSCNFs preserve 76.7% capacitance at current density of 10 A g−1 relative to 1 A g−1, however, only 66.3% for VCNFs. When adding initial vanadium loading to 15%, the rate performance is decreased in some cases, but the general capacitance is greater than other samples. Notably, increasing temperature can enhance rate performance, but impair capacitance, probably due the destructive pore structure.
Fig. 10a shows the Nyquist plots for PCNFs-800, SCNFs-800, VCNFs-800 and 15VSCNFs-800 at open-circuit potential. Nyquist plot consists of two parts. The semi-circle part at high frequency represents electron transfer rate, and the line part with a certain slope at low frequency stands for ion diffusion ability.50 The electrode series resistance of PCNFs-800 (35.6 Ω) is much higher than three others by comparing the semi-circle part, and the series resistance decreases slightly with the order of SCNFs-800 (1.8 Ω), VCNFs-800 (0.96 Ω) and 15VSCNFs-800 (0.48 Ω) (inserting plot in Fig. 10a), demonstrating vanadium and SWCNTs synergistically enhance CNFs conductivity by improving the graphene flake integrity. The ideal capacitor behaviour of SCNFs-800 is identified by the dramatically increase of angle from 45° to 80° after loading SWCNTs. Similarly, the slightly grow of slopes of SCNFs-800, VCNFs-800 and 15VSCNFs-800 may confirm that both SWCNTs and vanadium are helpful to build three-dimensional network among composite CNFs skeleton.
 | ||
| Fig. 10 (a) Nyquist plots of samples carbonized at 800 °C (the inserted graph in Nyquist plots is partially enlarged details at high frequency); (b) cyclic performance of 15VSCNFs-800 measured by galvanostatic charge–discharge method at current density of 8 A g−1 over 5000 cycles. | ||
GCD over 5000 cycles at current density of 8 A g−1 are performed to evaluate the cyclic performance of composite CNFs electrode. The best performance electrode 15VSCNFs-800 remain 94% of initial capacitance after 5000 cycles (Fig. 10b), suggesting impressive stability. Its charge efficiency (the ratio of charge and discharge amount) also remains 90.5% after cycling, indicating stable series resistance. Notably, this stable and low series resistance can increase energy efficiency without producing wasted heat. The similar charge and discharge curves at the beginning and final cycles also indicate the good stability of 15VSCNFs-800.
From the above discussions, high conductivity with lowest series resistance and good pseudo-capacitive behaviour are confirmed for 15VSCNFs-800, which supports the conclusion that the V–N–C and V–O–C bonds have formed inside graphite sheet fractures, and SWCNTs have contributed to build a unique capacitive structure, finally, this unique integrated structure enhance the stability and electrochemical performance of 15VSCNFs-800, resulting in superior supercapacitor.
4. Conclusions
We have synthesized ternary composite VSCNFs electrode with hierarchical structure through hybrid-electrospinning and carbonization technique. The composite VSCNFs have regular diameter distribution, stable carbon skeleton and enhanced graphitization due to the unique integrated structure formed through V–N–C, V–O–C and VO bonds by linking carbon skeleton together. 15VSCNFs-800 demonstrates the highest current response of 35 A g−1, the lowest series resistance (0.48 Ω) and quick ion transfer channel, specific capacitance (CGCD) of 479 F g−1 at current density of 1 A g−1 and good cycling stability with remaining 94% of initial capacitance after 5000 cycles at current density of 8 A g−1. In conclusion, the synergistic effect of vanadium and SWCNTs can greatly strengthen composite CNFs, tune pore structure and enhance electrochemical performance. Notably, this ternary composite VSCNFs electrode may also have the potential applications in water treatment and lithium batteries due to the widely use of vanadium in modifying electrodes.
Acknowledgements
This work was supported by national Natural Science Foundation of China (NSFC, no. 21377130 and 21177130). The authors would like to thank Dr Mingjie Li for his assistance in testing electrochemical performance.Notes and references
- S. Koohi-Kamali, V. V. Tyagi, N. A. Rahim, N. L. Panwar and H. Mokhlis, Renewable Sustainable Energy Rev., 2013, 25, 135 CrossRef PubMed.
- S. Chen, W. Xing, J. J. Duan, X. J. Hu and S. Z. Qiao, J. Mater. Chem. A, 2013, 1, 2941 CAS.
- E. Frackowiak and F. Beguin, Carbon, 2001, 39, 937 CrossRef CAS.
- A. Davies and A. P. Yu, Can. J. Chem. Eng., 2011, 89, 1342 CrossRef CAS PubMed.
- V. V. N. Obreja, Phys. E, 2008, 40, 2596 CrossRef CAS PubMed.
- C. Kim, J. Power Sources, 2005, 142, 382 CrossRef CAS PubMed.
- U. Fischer, R. Saliger, V. Bock, R. Petricevic and J. Fricke, J. Porous Mater., 1997, 4, 281 CrossRef CAS.
- Y. F. Yan, Q. L. Cheng, Z. J. Zhu, V. Pavlinek, P. Saha and C. Z. Li, J. Power Sources, 2013, 240, 544 CrossRef CAS PubMed.
- E. Frackowiak, K. Metenier, V. Bertagna and F. Beguin, Appl. Phys. Lett., 2000, 77, 2421 CrossRef CAS PubMed.
- P. Kossyrev, J. Power Sources, 2012, 201, 347 CrossRef CAS PubMed.
- Y. Q. Sun, Q. O. Wu and G. Q. Shi, Energy Environ. Sci., 2011, 4, 1113 CAS.
- M. Vangari, T. Pryor and L. Jiang, J. Energy Eng., 2013, 139, 72 CrossRef.
- F. Y. Zeng, Y. F. Kuang, N. S. Zhang, Z. Y. Huang, Y. Pan, Z. H. Hou, H. H. Zhou, C. L. Yan and O. G. Schmidt, J. Power Sources, 2014, 247, 396 CrossRef CAS PubMed.
- H. Jiang, P. S. Lee and C. Z. Li, Energy Environ. Sci., 2013, 6, 41 CAS.
- M. Endo, T. Hayashi, Y. A. Kim, M. Terrones and M. S. Dresselhaus, Philos. Trans. R. Soc., A, 2004, 362, 2223 CrossRef CAS PubMed.
- H. Q. Hou, J. J. Ge, J. Zeng, Q. Li, D. H. Reneker, A. Greiner and S. Z. D. Cheng, Chem. Mater., 2005, 17, 967 CrossRef CAS.
- M. Y. Ho, P. S. Khiew, D. Isa, T. K. Tan, W. S. Chiu and C. H. Chia, Nano, 2014, 9, 1430002, DOI:10.1142/S1793292014300023.
- X. J. Zhu, H. L. Dai, J. Hu, L. Ding and L. Jiang, J. Power Sources, 2012, 203, 243 CrossRef CAS PubMed.
- J. M. Miller, B. Dunn, T. D. Tran and R. W. Pekala, J. Electrochem. Soc., 1997, 144, L309 CrossRef CAS PubMed.
- A. Y. Shan, T. I. M. Ghazi and S. A. Rashid, Appl. Catal., A, 2010, 389, 1 CrossRef CAS PubMed.
- C. C. Xiang, M. Li, M. J. Zhi, A. Manivannan and N. Q. Wu, J. Mater. Chem., 2012, 22, 19161 RSC.
- W. T. Deng, X. B. Ji, Q. Y. Chen and C. E. Banks, RSC Adv., 2011, 1, 1171 RSC.
- A. Ghosh, E. J. Ra, M. Jin, H. K. Jeong, T. H. Kim, C. Biswas and Y. H. Lee, Adv. Funct. Mater., 2011, 21, 2541 CrossRef CAS PubMed.
- V. Aravindan, Y. L. Cheah, W. F. Mak, G. Wee, B. V. R. Chowdari and S. Madhavi, ChemPlusChem, 2012, 77, 570 CrossRef CAS PubMed.
- J. S. Im, S. W. Woo, M. J. Jung and Y. S. Lee, J. Colloid Interface Sci., 2008, 327, 115 CrossRef CAS PubMed.
- X. Pan, Y. Zhao, G. F. Ren and Z. Y. Fan, Chem. Commun., 2013, 49, 3943 RSC.
- H. W. Wang, H. Yi, X. Chen and X. F. Wang, J. Mater. Chem. A, 2014, 2, 1165 CAS.
- X. Chen, B. T. Zhao, Y. Cai, M. O. Tade and Z. P. Shao, Nanoscale, 2013, 5, 12589 RSC.
- K. L. Huang, X. G. Li, S. Q. Liu, N. Tan and L. Q. Chen, Renewable Energy, 2008, 33, 186 CrossRef CAS PubMed.
- B. H. Kim, C. H. Kim, K. S. Yang, A. Rahy and D. J. Yang, Electrochim. Acta, 2012, 83, 335 CrossRef CAS PubMed.
- X. W. Mao, T. A. Hatton and G. C. Rutledge, Curr. Org. Chem., 2013, 17, 1390 CrossRef CAS.
- J. Liu and W. X. Zhang, J. Appl. Polym. Sci., 2005, 97, 2047 CrossRef CAS PubMed.
- M. J. Yu, C. G. Wang, Y. J. Bai, Y. Xu and B. Zhu, J. Appl. Polym. Sci., 2008, 107, 1939 CrossRef CAS PubMed.
- D. H. Reneker, A. L. Yarin, H. Fong and S. Koombhongse, J. Appl. Phys., 2000, 87, 4531 CrossRef CAS PubMed.
- S. L. Shenoy, W. D. Bates, H. L. Frisch and G. E. Wnek, Polymer, 2005, 46, 3372 CrossRef CAS PubMed.
- Y. M. Shin, M. M. Hohman, M. P. Brenner and G. C. Rutledge, Polymer, 2001, 42, 9955 CrossRef CAS.
- J. J. Stanger, N. Tucker, M. Staiger, K. Kirwan, S. Coles, D. Jacobs and N. Larsen, AIP Conf. Proc., 2009, 1151, 118 CrossRef CAS PubMed.
- G. Lota, B. Grzyb, H. Machnikowska, J. Machnikowski and E. Frackowiak, Chem. Phys. Lett., 2005, 404, 53 CrossRef CAS PubMed.
- Z. Y. Zhang, X. H. Li, C. H. Wang, S. W. Fu, Y. C. Liu and C. L. Shao, Macromol. Mater. Eng., 2009, 294, 673 CrossRef CAS PubMed.
- D. S. Yuan, J. X. Chen, S. X. Tan, N. N. Xia and Y. L. Liu, Electrochem. Commun., 2009, 11, 1191 CrossRef CAS PubMed.
- X. Zhao, W. Li and S. X. Liu, Mater. Lett., 2013, 107, 5 CrossRef CAS PubMed.
- E. G. Ferrer and E. J. Baran, J. Electron Spectrosc. Relat. Phenom., 1991, 57, 189 CrossRef CAS.
- M. S. Dresselhaus, A. Jorio, A. G. Souza and R. Saito, Philos. Trans. R. Soc., A, 2010, 368, 5355 CrossRef CAS PubMed.
- H. T. Niu, J. Zhang, Z. L. Xie, X. G. Wang and T. Lin, Carbon, 2011, 49, 2380 CrossRef CAS PubMed.
- G. D. Stucky, D. A. Mathews, M. Klasson, J. Hedman and C. Nordling, J. Am. Chem. Soc., 1972, 94, 8009 CrossRef CAS.
- C. D. Batich and D. S. Donald, J. Am. Chem. Soc., 1984, 106, 2758 CrossRef CAS.
- M. D. Antonik, R. J. Lad and T. M. Christensen, Surf. Interface Anal., 1996, 24, 681 CrossRef CAS.
- E. J. Baran, A. H. Jubert and E. G. Ferrer, J. Raman Spectrosc., 1992, 23, 489 CrossRef CAS PubMed.
- E. J. Baran and K. H. Lii, Z. Naturforsch., B: J. Chem. Sci., 2003, 58, 485 CAS.
- P. L. Taberna, P. Simon and J. F. Fauvarque, J. Electrochem. Soc., 2003, 150, A292 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Table of transitional pre-oxidization procedure, SEM images captured at low magnification, N2 sorption isotherm of samples carbonized at 900 °C, nanofiber diameter distributions, FT-IR spectrum, CV and GCD curves, Nyquist plots of samples carbonized at 900 °C, TEM images, electrode digital images. See DOI: 10.1039/c5ra02237a |
| This journal is © The Royal Society of Chemistry 2015 |