
Apigenin mediated gold nanoparticle synthesis and their anti-cancer effect on human epidermoid carcinoma (A431) cells†
Indra Rajendrana,
Harini Dhandapania,
Rajaram Anantanarayananb and
Rama Rajaram*a
aDepartment of Biochemistry, Central Leather Research Institute, Adyar, Chennai – 600 020, India. E-mail: rajaram.rama@gmail.com; Tel: +91-44-24437177
bDepartment of Biophysics, Central Leather Research Institute, Adyar, Chennai – 600 020, India
First published on 2nd June 2015
Abstract
We report that the flavonoid, apigenin (4′,5,7,-trihydroxyflavone) is able to form apigenin linked gold nanoparticles (ap-AuNPs) at RT with apigenin itself acting as the stabilizing agent. The synthesized ap-AuNPs have been characterised by UV-Visible spectroscopy, HR-TEM, DLS, FTIR and TGA analyses. The biocompatible nature of ap-AuNPs is shown by their non-toxicity towards normal epidermoid cells (HaCat). Additionally, they are shown to exhibit anti-cancer activity towards epidermoid squamous carcinoma cells (A431). The uptake of ap-AuNPs into A431 cells is seen by TEM. Apoptosis induced by ap-AuNPs in these cells is observed through EtBr/AO and DAPI staining. Flow cytometric results also reveal the occurrence of early apoptotic cells, late apoptotic cells and the presence of sub-G1 population. DNA prepared from ap-AuNPs treated A-431 cells reveals a ladder like pattern in agarose gel electrophoresis indicating oligonucleosomal cleavage which is a hallmark of apoptosis. The ap-AuNPs also inhibit angiogenesis as shown by Chick chorioallantoic membrane (CAM) assay. Taking the results together, we believe that ap-AuNPs show promise in treatment of skin cancer. The ap-AuNPs also have cytotoxic potential against the human cervical squamous cell carcinoma cell line SiHa.
Introduction
The incidence of skin cancer has been increasing during the last decade.1 Overexposure to solar UV radiation and chemicals such as arsenic and nickel are the major etiological factors for the development of malignancy.2–4 Therefore, there is a need to develop novel therapeutic agents which preferably target cancer cells leaving normal cells unaffected. Gold nanoparticles (AuNPs) have attracted considerable attention due to their possible applications in cancer treatment, in drug delivery, as biosensors, in photo thermal therapy and in imaging.5–10 AuNPs have been synthesized by reducing Au3+ ions employing chemical reductants such as citrate, CTAB, etc., and the shape and size of the particles are tunable by adjusting the conditions employed.11–13 Applications in biomedicine are however limited when the capping and stabilizing agents used have toxic effects in biological systems.11,13 For example, citrate reduced AuNPs are found to be toxic towards normal human dermal microvascular endothelial cells (HDMEC) and human cerebral microvascular endothelial cells (hCMEC).13 The synthesis of AuNPs using plant extracts and phytochemicals may prove advantageous over the AuNPs made via chemical capping.14–16 Phytochemicals like (−) epigallocatechin-3-gallate (EGCG), resveratrol, tannins etc., have all been used for forming colloidal gold from ionic solutions of gold.16–18The flavones which are one of the secondary metabolites of plants, exhibit antioxidant, antimicrobial, anti-proliferative and anti-carcinogenic properties.19,20 Flavonoids like apigenin, quercetin, genistein etc., have been shown to exhibit anti-cancer activity while being nontoxic to normal cells.20 Some of the flavones that have been shown to reduce Au3+ ions to form AuNPs are apiin, dihydromyricetin, quercetin, tannic acid and rutin. The –OH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups present are reported to aid in the reduction of Au3+ and in the stabilization of the AuNPs formed.16,21–24
O groups present are reported to aid in the reduction of Au3+ and in the stabilization of the AuNPs formed.16,21–24
Apigenin is a flavonoid (4′,5,7,-trihydroxyflavone), derived from edible fruits and vegetables including parsley, onions, oranges, tea, chamomile, wheat sprouts and some seasonings.25 It is non-mutagenic and possesses antimicrobial, antiviral and anti-carcinogenic activities.26,27 When prostate cancer cells (22Rv1) are exposed to apigenin, enhanced ROS generation, activation of wild-type p53 and induction of apoptosis have been observed.28 Skin carcinoma induced by 12-O-tetradecanoylphorbol-13-acetate (TPA) in mice model has been shown to be inhibited by topical application of 20 μmol kg−1 body weight of apigenin.27 It also inhibits the transcription of vascular endothelial growth factor (VEGF) in ovarian cells and thus inhibits tumour growth and tumour angiogenesis in mice.29,30 The concomitant antitumor activity of apigenin and ABT-263 (Bcl-2 inhibitor) is found to be enhanced both in vitro and in vivo in human colon cancer cells.31 Recent reports indicate that apigenin suppresses proliferation and migration and induces apoptosis of T24 bladder cancer cells and breast cancer (T47D and MDA-MB-231) cells.32,33 In human skin malignant melanoma (A375) and lung carcinoma cell lines (A549), apigenin induces apoptosis at 80 and 90 μg mL−1 respectively by modulating the mitochondrial oxidative phosphorylation system.34
The effect of flavonoids is reported to be enhanced when they are in nano-formulations.35 The possible use of flavonoids in the treatment of several cancers also suggest the use of ap-AuNPs in treating skin cancer.14,36 However it is not known whether apigenin can reduce Au3+ to form stable gold nanoparticles and if they have any effect on carcinogenic epidermoid cells. Here we report that apigenin conjugated to gold nanoparticles (ap-AuNPs) are formed when apigenin reacts with Au3+ under appropriate conditions. The ap-AuNPs are also found to exhibit toxicity towards A431 (epidermoid squamous cell carcinoma) cells while being non-toxic towards normal epidermoid cells (HaCat). Further, the cytotoxic effect of these ap-AuNPs on another squamous cell line (SiHa) is also demonstrated.
Results and discussion
Synthesis and formation of ap-AuNPs
The formation of ap-AuNPs has been investigated using different molar ratios of apigenin to HAuCl4 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:5) and at different pH values of apigenin (8–11). Mixing apigenin and HAuCl4 solutions at neutral pH do not result in the formation of particles. Formation of ap-AuNPs occurs at RT when the pH of apigenin is increased as observed through the color change from straw yellow to burgundy red. This has also been monitored using a spectrophotometer (Fig. 1). When the pH of apigenin is increased to 8, a small surface plasmon resonance (SPR) peak at 540 nm is observed indicating ap-AuNPs formation, but the particles formed are found to be unstable. As the pH is increased to 9, a new peak at 750 nm is observed in addition to the peak at 540 nm (Fig. 1a). This may indicate the formation of nanoparticles which are of different shapes. Increase in intensity as well as narrowing of the SPR peak at 540 nm is observed when the pH of apigenin is increased to 10 and this generally is a characteristic of monodispersed nanoparticles. Since the pKa value of apigenin as determined by HPLC method is ∼10 the complete solubility of apigenin at this pH may be leading to the reduction of Au3+ to Au0 and hence the formation of monodispersed AuNPs.37 On increasing the pH of apigenin to 11, a turbid red coloured colloidal solution is formed and the SPR peak shifts to 560 nm. The particles are also found to settle down indicating that they are unstable. The results indicate that the pH of apigenin solution is crucial for the formation of uniformly sized nanoparticles. The pH dependency of AuNPs formation has also been observed with other flavones like dihydromyricetin and curcumin when they are the reductants.22,38 Since, ap-AuNPs prepared with apigenin at pH 10 only are found to be monodispersed, further characterisations have been performed on these samples.
:1 to 1:5) and at different pH values of apigenin (8–11). Mixing apigenin and HAuCl4 solutions at neutral pH do not result in the formation of particles. Formation of ap-AuNPs occurs at RT when the pH of apigenin is increased as observed through the color change from straw yellow to burgundy red. This has also been monitored using a spectrophotometer (Fig. 1). When the pH of apigenin is increased to 8, a small surface plasmon resonance (SPR) peak at 540 nm is observed indicating ap-AuNPs formation, but the particles formed are found to be unstable. As the pH is increased to 9, a new peak at 750 nm is observed in addition to the peak at 540 nm (Fig. 1a). This may indicate the formation of nanoparticles which are of different shapes. Increase in intensity as well as narrowing of the SPR peak at 540 nm is observed when the pH of apigenin is increased to 10 and this generally is a characteristic of monodispersed nanoparticles. Since the pKa value of apigenin as determined by HPLC method is ∼10 the complete solubility of apigenin at this pH may be leading to the reduction of Au3+ to Au0 and hence the formation of monodispersed AuNPs.37 On increasing the pH of apigenin to 11, a turbid red coloured colloidal solution is formed and the SPR peak shifts to 560 nm. The particles are also found to settle down indicating that they are unstable. The results indicate that the pH of apigenin solution is crucial for the formation of uniformly sized nanoparticles. The pH dependency of AuNPs formation has also been observed with other flavones like dihydromyricetin and curcumin when they are the reductants.22,38 Since, ap-AuNPs prepared with apigenin at pH 10 only are found to be monodispersed, further characterisations have been performed on these samples.
 | ||
| Fig. 1 UV-Visible spectra of the ap-AuNPs formed at different conditions of synthesis (a) ap-AuNPs formed at different pHs of apigenin (0.3 mM of apigenin and 1.2 mM of HAuCl4 is used for the synthesis); inset picture shows burgundy red color of ap-AuNPs formed at pH 10 (b) ap-AuNPs formed at different molar ratios of apigenin to HAuCl4 reacted at pH 10 (concentration of apigenin has been kept constant at 0.3 mM and the concentration of HAuCl4 ranges from 0.3 to 1.5 mM) and (c) kinetics of ap-AuNPs formation. | ||
In order to optimise the conditions for synthesis, reaction of apigenin with HAuCl4 at different molar ratios (1:1 to 1:5) has been carried out. On increasing the concentration of HAuCl4, the intensity of SPR peak at 540 nm also increases and at a molar ratio of 1:4, it is found to be the maximum (Fig. 1b). When the molar ratio is increased to 1:5, a shift in the SPR peak is observed (590 nm) and the peak is also broad indicating that the particles are not of uniform size. A small and clear shift in SPR towards longer wavelength and decrease in the bandwidth with increasing ratio of apigenin to HAuCl4 from 1:1 to 1:4 and pH of apigenin from 8–10 indicates that uniform particles are formed at the saturating concentrations than at lower concentrations (Fig. S1a and S1b†). Thus, a molar ratio of apigenin to HAuCl4 of 1:4 (0.3 mM of apigenin: 1.2 mM of HAuCl4) and a pH of apigenin at 10 are found to be the optimum parameters for the synthesis of monodispersed ap-AuNPs. The UV-Visible spectroscopy method has been used as a conventional method for qualitatively determining the yield of AuNPs. A SPR value of around 1.7 obtained for ap-AuNPs (when 1.2 mM of HAuCl4 is used) indicates that the yield is similar to that of AuNPs formed when citrate is used as the reductant.39
Characterisation of ap-AuNPs
 | ||
| Fig. 2 Characterization of ap-AuNPs formed at pH 10 (a) low magnification HR-TEM image with inset showing the size distribution of ap-AuNPs (b) a single particle of ap-AuNPs depicting lattice fringes (c) SAED pattern depicting the lattice planes of Au in ap-AuNPs (d) X-ray diffraction pattern illustrating crystalline nature of the ap-AuNPs (e) size of ap-AuNPs measured by DLS (f) stability of ap-AuNPs measured by zeta potential (g) FTIR spectrum of apigenin and ap-AuNPs (h) TGA showing the different phases of weight loss of the reductant apigenin on the ap-AuNps. | ||
O stretching vibrations appear in all the spectra indicating that the keto group remains unaltered at the end of the reaction. The CCC ring vibrations represented by the band at 1020 cm−1 and the alkyl ring vibrations associated with the band at 2924 cm−1 can be observed in the ap-AuNPs spectra indicating the presence of apigenin on the ap-AuNPs formed.42,43 These results show the involvement of the polar hydroxyl groups of apigenin in reducing and capping the ap-AuNPs.Mechanism of ap-AuNPs formation
Fig. 1c, illustrates the kinetics of the formation of ap-AuNPs when apigenin solution at pH 10 is used for synthesis. During ap-AuNPs formation, initially a broad absorption spectrum is obtained and as the reaction progresses, the SPR centred at 540 nm becomes narrower. Burgundy red coloured ap-AuNPs are obtained within 10–20 min of the reaction, and maximum SPR intensity is obtained after 3 h of reaction. The gradual narrowing of the peak and the uniform spherical particles obtained finally indicate that the nanoparticles might have formed through a process of temporal evolution where large sized particles are first formed which then get cleaved to smaller sized particles.22 The HR-TEM images indicate the presence of monodispersed spherical particles.At a pH of 9, besides the SPR peak at 540 nm, another peak at 750 nm is observed indicating the formation of different sized or shaped particles. This is reflected in the HR-TEM image where a mixture of spherical, triangular, hexagonal and oblong shaped particles with a broad size distribution is observed (Fig. S2†). Similar observations were made during the synthesis of AuNPs using fulvic acid where a decrease in pH leads to the formation of different sized particles while at higher pH monodispersed particles are obtained.41 Since the deprotonated hydroxyl groups of apigenin are assumed to reduce Au3+, and as apigenin at pH 9 is found to have only two hydroxyl ions when compared to that at pH 10 (3 hydroxyl ions) it can be inferred that the polydispersed particles produced at pH 9 might be due to the inability of apigenin at this pH to completely reduce Au3+. Thus the extent of deprotonation of apigenin is found to determine the size and shape of ap-AuNPs formed.
Uptake studies
The uptake of ap-AuNPs by A431 cells has been observed through transmission electron microscopy (TEM). The representative images of the uptake of ap-AuNPs by A431 cells after a treatment period of 48 h are presented in Fig. 3. Internalization of ap-AuNPs into the sub-cellular organelles is observed. The mode of internalization may be by the endocytosis pathway and the absence of aggregation of ap-AuNPs inside the cell reflects a high degree of stability. Citrate reduced AuNPs are also reported to undergo such an endocytotic fate in macrophage cells.45 Localization of AuNPs inside mitochondria and other organelles has also been observed in HeLa and MCF-7 cells.46 | ||
| Fig. 3 Uptake of ap-AuNPs in A431 cells as seen by transmission electron microscopy (a) internalization of ap-AuNPs (b) magnified image showing dispersed ap-AuNPs sequestered in phagolysosomes. | ||
Biocompatibility and cytotoxicity studies
Biocompatibility and cytotoxicity studies have been carried out with ap-AuNPs prepared at pH 10 and molar ratio of 1:4.
| Sample | % of haemolysis |
|---|---|
| Positive control – water | 100 ± 0.0 |
| Negative control – PBS | 1.9 ± 0.2 |
| ap-AuNPs 50 μg mL−1 | 1.0 ± 0.0 |
| ap-AuNPs 100 μg mL−1 | 1.0 ± 0.1 |
| ap-AuNPs 150 μg mL−1 | 1.7 ± 0.2 |
| ap-AuNPs 200 μg mL−1 | 2.6 ± 0.1 |
 | ||
| Fig. 4 Viability assay measured using MTT. The effect of ap-AuNPs on (a) HaCat cells (b) A431 cells (c) effect of ct-AuNPs on A431 cells and (d) effect of free apigenin on A431 cells. The values represent mean ± SD of three independent experiments. *p < 0.05, **p < 0.01 and ***p < 0.001 vs. control. | ||
In order to find out whether the toxicity caused by ap-AuNPs is due to the reductant apigenin, viability assay was conducted with free apigenin at concentrations corresponding to that present on ap-AuNPs. The results show that there is no significant decrease in viability on treatment with 20 μg mL−1 of free apigenin up to 72 h (Fig. 4d). A significant decrease is observed with 40 μg mL−1 of free apigenin after 48 and 72 h and the viability percentages are found to be 71.0 ± 3.5% and 65.5 ± 2.1% respectively. It is to be noted that in the presence of 50 μg mL−1 of ap-AuNPs (which contains only 10 μg mL−1 of bound apigenin), similar viability values are obtained, indicating that almost a four-fold increase in capacity to kill the cancer cells is realized by the ap-AuNPs conjugation.
The effect of ap-AuNPs on SiHa cells has also been studied. The results show a concentration and time dependent decrease in viability. On treatment with 200 μg mL−1 of ap-AuNPs for 24 h, the viability is found to be 52.7 ± 2.6%, which further decreases to 44.2 ± 3.1% and 31.8 ± 1.1% after 48 h and 72 h respectively (Fig. S5a†). Gallic acid conjugated gold nanoparticles (150 μM) are reported to induce cell death in SiHa cells.48 The viability results are comparable to that of A431 cells indicating that the ap-AuNPs exhibit comparable cytotoxicity towards these two types of epidermoid squamous carcinoma cell lines.
Microscopy analysis
 | ||
Fig. 5 Fluorescence micrographs using EtBr/AO staining of (a) HaCat cells and (b) A431 cells on treatment with ap-AuNPs. The morphological changes are indicated as early apoptotic ( ), late apoptotic ( ), late apoptotic ( ) and necrotic cells ( ) and necrotic cells ( ) (c) fluorescence micrographs using DAPI staining. The morphological changes are indicated as apoptotic bodies ( ) (c) fluorescence micrographs using DAPI staining. The morphological changes are indicated as apoptotic bodies ( ), chromatin condensation ( ), chromatin condensation ( ) and chromatin condensation with membrane damage ( ) and chromatin condensation with membrane damage ( ). ). | ||
The nuclear damage is analysed through DAPI staining. DAPI is a fluorescent chromophore, which binds to double stranded DNA. Fig. 5c shows control cells with uniform round nuclei indicating that they are viable. When 200 μg mL−1 of ap-AuNPs are present, cells show crescent shaped nuclei and chromatin condensation with membrane damage, which is characteristic of apoptosis. Thus, these results suggest that the cell death brought about by ap-AuNPs may be through apoptosis.
The ap-AuNPs induced cell death on SiHa cells has also been analysed through EtBr/AO staining. Fig. S5b.† shows the membrane damage and reddish-orange stained cells in the presence of 100 μg mL−1 of ap-AuNPs. When treated with 200 μg mL−1 of ap-AuNPs, a few cells are reddish-orange in color indicating late apoptotic phase and comparatively more number of cells are with red nuclei indicating necrosis.
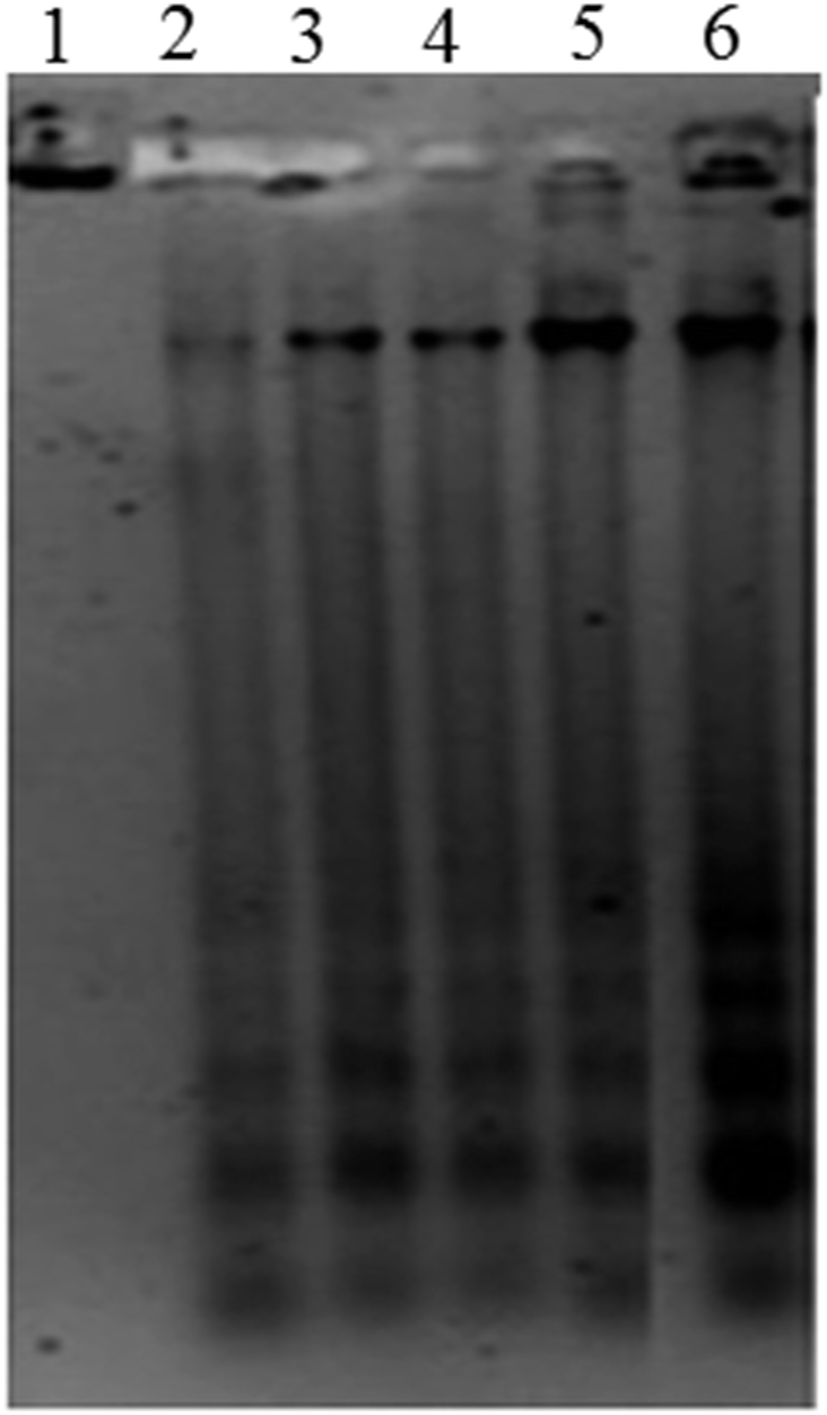 | ||
| Fig. 6 DNA fragmentation assessed using 1% of agarose gel electrophoresis. Genomic DNA extracted from A431 cells without (control) and after treatment with various concentration of ap-AuNPs for 48 h. Lane 1: control, Lane 2: ap-AuNPs 25 μg mL−1, Lane 3: ap-AuNPs 50 μg mL−1, Lane 4: ap-AuNPs 100 μg mL−1, Lane 5: ap-AuNPs 150 μg mL−1, Lane 6: ap-AuNPs 200 μg mL−1. | ||
Flowcytometry analysis
Phosphatidylserine (PS) is present on the cytoplasmic surface of the cell membrane in viable cells. When the cells undergo apoptosis, PS is translocated from the inner to the outer leaflet of the plasma membrane. Annexin V is a 35–36 kDa Ca2+ dependent phospholipid-binding protein that has high affinity for PS and hence translocation of PS can be analysed using fluorescein isothiocyanate (FITC) conjugated to annexin V through flow cytometry.50 Dual staining of treated cells with annexin V-FITC and PI analysed through flow cytometry is shown in Fig. 7a. Representative dot plot in the lower left quadrant denotes viable cells while the lower right and upper right quadrant populations represent early and late apoptotic cells respectively. Upper left quadrant population of cells stained only with PI denotes necrotic cells. Thus the ap-AuNPs induced cell death of A431 cells in a concentration and time dependent manner is clearly observed. The percentage of viable cells which is found to be 75.4 ± 3.2% and 64.3 ± 1.3% on treatment with 150 and 200 μg mL−1 respectively for 24 h, decreases to 47.7 ± 2.2% and 34.9 ± 2.1% after 48 h. Corresponding increase in early apoptotic cell population as 12.5 ± 1.5% and 21.1 ± 1.6% respectively is observed after 24 h which further increases to 29.6 ± 3.8% and 42.5 ± 2.8% after 48 h of treatment. These results confirm that the cells undergo apoptosis on treatment with ap-AuNPs. Recently, it has been shown that Cajanus cajan reduced AuNPs induce early apoptosis of Hep-G2 cells which has been identified through annexin V FITC analysis.51 | ||
| Fig. 7 Flow cytometry analyses of A431 cells on treatment with different concentration of ap-AuNPs. (a) Annexin-V-FITC binding and PI uptake (b) cell cycle analysis using PI fluorescence. Dot plots and histogram plots are representative of three independent experiments. | ||
Cell cycle comprises four different phases (G1 phase, S phase, G2 phase and mitosis) and two checkpoints (G0/G1 and G2/M checkpoints) which ensures that DNA damage is not transmitted to daughter cells.52,53 Cells treated with ap-AuNPs show gradual decrease in G0/G1 phase and corresponding increases in cell population in S-phase and Sub-G1 fraction after 24 h (Fig. 7b). An increasing S-phase population from 18.1 ± 1.5% (control) to 27.3 ± 0.7% (150 μg mL−1 of ap-AuNPs) and 29.3 ± 2.5 (200 μg mL−1 of ap-AuNPs) indicates cell cycle arrest in S-phase. The percentage of Sub-G1 population is 1.8 ± 0.6% in control, whereas after treatment with 50, 100 and 150 μg mL−1 of ap-AuNPs for 24 h, the values are 5.6 ± 1.5%, 10.1 ± 0.4% and 13.8 ± 2.7% respectively. For the same time period, the value further increases to 21.7 ± 3.2% in the presence of 200 μg mL−1 of ap-AuNPs. The Sub-G1 population is as high as 31.1 ± 4.3% after 48 h of treatment with 200 μg mL−1 of ap-AuNPs, with a concomitant decrease in G0/G1 (29.8 ± 2.3%) and G2/M (12.6 ± 1.6%) phases. The increase in Sub-G1 population with increase in ap-AuNPs concentration and treatment period also indicates the apoptotic mode of cell death. Thus, the characteristic morphological features of apoptosis observed though EtBr/AO and DAPI staining are confirmed through DNA fragmentation and flow-cytometric analysis. AuNPs have been reported to induce apoptosis of several cancer cells via S phase and Sub-G1 arrest.50,53,54
Anti-angiogenic potential exerted by ap-AuNPs (CAM assay)
To test if ap-AuNPs possess anti-angiogenic activity, in vivo chick embryo chorioallantoic membrane (CAM) assay has been performed. CAMs were treated with different concentrations of ap-AuNPs for 48 h. The ap-AuNPs at a concentration of 200 μg mL−1 inhibits the formation of new blood vessels and sub branches when compared to that of control and VEGF treated chick embryos (Fig. 8). Though the ability of pure apigenin (20 μM) to inhibit angiogenesis in prostate cancer cells through HIF and VEGF expression has been demonstrated30 that apigenin capped AuNPs (ap-AuNPs) can also limit the growth of new blood capillaries is demonstrated in this study. The results on the ability of ap-AuNPs to inhibit the development of blood vessels indicate their anti-angiogenic property implying their value as agents for skin cancer treatment. | ||
| Fig. 8 The anti-angiogenic potential of ap-AuNPs analyzed using CAM assay. Arrows point to the presence of blood vessels (control and VEGF) and inhibition of existing blood vessels in ap-AuNPs treated CAM. | ||
Materials and methods
Hydrogen tetrachloroaurate (HAuCl4·3H2O, >99.9%), apigenin, Dulbecco's Modified Eagle's Medium (DMEM), phosphate buffered saline (PBS), sodium bicarbonate, streptomycin, gentamicin, penicillin, amphotericin B, trypsin–EDTA, ethidium bromide (EtBr), acridine orange (AO), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 4′-6-diamidino-2-phenylindole (DAPI), agarose, propidium iodide (PI), DNase free RNase, proteinase K, 4-(2-hydroxyethyl)-1-piperazineethane sulfonic acid (HEPES), ethylenediaminetetracetic acid (EDTA) and dimethylsulphoxide (DMSO) were purchased from Sigma Aldrich (USA). Fetal Bovine Serum (FBS) was purchased from GIBCO (USA). Ammonium acetate, Nonidet P-40, potassium carbonate (K2CO3), potassium bromide (KBr), tris hydrochloride and formaldehyde were obtained from Merck (USA). Annexin-V-FITC apoptosis kit was obtained from BD Bioscience, USA. Dodecenyl succinic acid (DDSA), epon 812 resin, glutaraldehyde, lead citrate, nadic methyl anhydride (NMA), sodium cacodylate buffer and uranyl acetate were bought from EM Sciences, USA. MilliQ water (conductivity of 18 mΩ cm−1) was autoclaved and filter-sterilised using 0.22 μm filters. Cells (A431, HaCat and SiHa) were obtained from the National Centre for Cell Science, Pune, India.Synthesis of apigenin conjugated gold nanopaticles (ap-AuNPs)
A stock solution of 10 mM apigenin was prepared by dissolving in 100% DMSO. Aqueous stock solution of HAuCl4 (10 mM) and K2CO3 (150 mM) were prepared in filtered MilliQ water. The pH of the apigenin solution (0.3 mM) was adjusted to 10 using K2CO3. Then, 1.2 mM of HAuCl4 was added drop wise while mixing the solution of apigenin at RT. The reduction of Au3+ to Au0 was monitored spectrophotometrically (280–900 nm) using a multimode plate reader (Tecan Infinite M 200). Reduction was also assessed using different concentrations of HAuCl4 (0.3 to 1.6 mM) and at different pH values (8–11). The resultant ap-AuNPs solution was allowed to age at RT for 4 days and centrifuged at 15000×g for 10 min at a temperature of 15 °C. The pellet was resuspended with MilliQ water and washed thrice to eliminate unreacted apigenin and HAuCl4. Finally, the pellet was resuspended in MilliQ water and then used for further characterisation. Citrate reduced gold nanoparticles (ct-AuNPs) were also prepared by reacting sodium citrate and HAuCl4 in the ratio of 1:6.8.55 This has been used as a control for comparing the cytotoxicity of ap-AuNPs.
Characterisation of ap-AuNPs
Biocompatibility of ap-AuNPs: haemolysis assay
Haemocompatibility studies were performed after getting approval from the Institutional Review Board (IRB) of Central Leather Research Institute (CLRI), Chennai, India. Blood was collected from healthy donors in a heparinised tube and red blood cells (RBCs) were separated by centrifugation. The isolated RBCs were washed four times with Dulbecco's phosphate buffered saline (PBS) at 400×g for 5 min, and then diluted 10 times with PBS. To 0.2 mL of RBCs, 0.8 mL of different concentrations of ap-AuNPs in PBS were added and incubated for 4 h at 37 °C. The cells were then vortexed and centrifuged at 400×g for 10 min. The absorbance of haemoglobin was measured at 577 nm in 200 μL of the supernatant with reference wavelength at 655 nm in a multimode plate reader (Tecan Infinite M 200). Water and PBS served as positive and negative controls respectively. The percentage of haemolysis was calculated using the formula as below
Cell culture
Cells were grown in DMEM medium containing 10% FBS, 100 μg mL−1 streptomycin, 100 μg mL−1 gentamycin, 100 U mL−1 penicillin and 2.5 μg mL−1 amphotericin B. Cells were trypsinized at 70–80% confluency using 0.25% trypsin–EDTA and seeded for the experiments. The cells were allowed to attach overnight and then treated with ap-AuNPs while untreated cells served as control. The cells were maintained at 37 °C with 5% CO2 in a CO2 incubator (Binder, Germany).Uptake studies
For TEM analysis, 1 × 106 cells were grown in a T25 flask and treated with 100 μg mL−1 of ap-AuNPs for 48 h. After incubation, the cells were washed thrice with PBS and trypsinized. The cells were then fixed with 2.5% of glutaraldehyde prepared in 0.1 M sodium cacodylate buffer (pH 7.4) for 4 h. The pellet was then centrifuged and washed with the same buffer thrice for 10 min each. They were then post fixed in 0.1% osmium tetroxide prepared in the same buffer for 2 h at 8 °C and further washed as above. The cells were pelleted through centrifugation after each step and further dehydrated through a graded series of acetone 30%, 50%, 70%, 80%, and 90% for 10 min. It was then treated with 100% acetone twice for 10 min, each time followed by propylene oxide treatment for 10 min. The pellet was then infiltrated with resin mixture consisting of Epon 812 resin, DDSA and NMA starting with 25%, 50% and 75% for 2 h and 100% overnight. It was then embedded in the same resin mixture with catalyst (DMP 30) in “easymoulds” at 60 °C for 48 h. Ultra thin sections from the resin block were cut and stained with saturated solutions of uranyl acetate and lead citrate. After air drying, the sections were visualized in Jeol JEM 1400 transmission electron microscope at 80 kV and micrographs acquired using Olympus Keen view CCD camera.MTT viability assay
The viability of HaCat, A431 and SiHa cells on exposure to ap-AuNPs were determined by MTT assay. Cells (1 × 104) were treated with various concentrations of ap-AuNPs for 0–72 h. A431 cells were also treated with apigenin, ct-AuNPs and compared with that of ap-AuNPs. They were then washed with sterile PBS and incubated with MTT (0.5 mg mL−1) solution for 4 h at 37 °C. The formazan product formed was dissolved in 200 μl of DMSO and the absorbance was measured at 570 nm using a reference wavelength of 630 nm in a multimode plate reader (Tecan Infinite M 200). Viability test was also conducted on cells treated with 10, 20, 30 and 40 μg mL−1 of free apigenin. Experiments were conducted in triplicate and the percentage of viable cells was calculated with respect to control.Microscopy studies
000 × g for 20 min and dissolved in Tris–EDTA buffer. The DNA samples were subjected to electrophoresis on 1% agarose gel and the image was documented using Gel Documentation system (Bio-Rad, USA).For cell cycle analysis, treated cells were trypsinized, fixed with 70% ethanol and stored at −20 °C overnight. They were then suspended in 0.5 mL of sterile PBS containing 50 μg mL−1 of DNase free RNase and 50 μg mL−1 of PI at RT in the dark for 20 min. From the fluorescence measured at 488 nm using flow cytometer, the cells present in the various phases were analysed using Cell Quest Pro software.
Conclusion
Our study on the synthesis of ap-AuNPs using apigenin has revealed that Au3+ can be reduced by apigenin at a pH of 10 and at RT forming highly stable and spherical ap-AuNPs. These ap-AuNPs are biocompatible towards normal epidermoid cells (HaCat) while inducing apoptosis of A431 and SiHa cells at the concentrations used here. The ap-AuNPs also exhibit good anti-angiogenic property. Hence, ap-AuNPs can be considered as a promising candidate for use in skin cancer treatment.Conflict of interest
The authors have no conflict of interest.Acknowledgements
The authors thank the Director, CLRI, Chennai, India for providing the necessary facilities and support. The work was carried out with financial assistance through the Council of Scientific and Industrial Research, India, XII five year plan project (Nano SHE-BSC 0112). The authors thank the staff of TEM facilities of the National Centre for Nanoscience and Nanotechnology, University of Madras and the Cancer Institute, Chennai.References
- R. S. Laishram, A. Banerjee, P. Punyabati and L. D. C. Sharma, J. Pak. Assoc. Derma., 2010, 20, 128–132 Search PubMed.
- M. R. Karagas, T. A. Stukel, J. S. Morris, T. D. Tosteson, J. E. Weiss, S. K. Spencer and E. R. Greenberg, Am. J. Epidemiol., 2001, 153, 559–565 CrossRef CAS PubMed.
- V. Peharda, F. Gruber, M. Kastelan, L. P. Massari, M. Saftic, L. Cabrijan and G. Zamolo, Coll. Antropol., 2007, 31, 87–90 Search PubMed.
- F. Gruber, V. Peharda, M. Kastelan and I. Brajac, Acta Dermatovenerol. Croat., 2007, 15, 191–198 Search PubMed.
- A. Llevot and D. Astruc, Chem. Soc. Rev., 2012, 41, 242–257 RSC.
- R. M. Navalakhe and T. D. Nandedkar, Indian J. Exp. Biol., 2007, 45, 160–165 CAS.
- P. M. Tiwari, K. Vig, V. A. Dennis and S. R. Singh, Nanomaterials, 2011, 1, 31–63 CrossRef CAS PubMed.
- E. E. Bedford, J. Spadavecchia, C. M. Pradier and F. X. Gu, Macromol. Biosci., 2012, 12, 724–739 CrossRef CAS PubMed.
- X. Q. Zhao, T. X. Wang, W. Liu, C. D. Wang, D. Wang, T. Shang, L. Shen and L. Ren, J. Mater. Chem., 2011, 21, 7240–7247 RSC.
- C. Pache, N. L. Bocchio, A. Bouwens, M. Villiger, C. Berclaz, J. Goulley, M. I. Gibson, C. Santschi and T. Lasser, Opt. Express, 2012, 20, 21385–21399 CrossRef CAS PubMed.
- S. Vijayakumar and S. Ganesan, J. Nanomater., 2012, 1–9 CrossRef PubMed.
- S. Sasidharan, A. Jayasree, S. Fazal, M. Koyakutty, S. V. Nair and D. Menon, Biomater. Sci., 2013, 1, 294–305 RSC.
- C. Freese, C. Uboldi, M. I. Gibson, R. E. Unger, B. B. Weksler, I. A. Romero, P. O. Couraud and C. J. Kirkpatrick, Part. Fibre Toxicol., 2012, 9, 1–11 CrossRef PubMed.
- D. Raghunandan, S. Basavaraja, B. Mahesh, S. Balaji, S. Y. Manjunath and A. Venkataraman, Nanobiotechnology, 2009, 5, 34–41 CrossRef CAS.
- K. B. Narayanan and N. Sathivel, Mater. Lett., 2008, 62, 4588–4590 CrossRef CAS PubMed.
- S. K. Nune, N. Chanda, R. Shukla, K. Katti, R. R. Kulkarni, S. Thilakavathy, S. Mekapothula, R. Kannan and K. V. Katti, J. Mater. Chem., 2009, 19, 2912–2920 RSC.
- R. K. Mohanty, S. Thennarasu and A. B. Mandal, Colloids Surf., B, 2014, 114, 138–143 CrossRef CAS PubMed.
- S. A. Aromal and D. Philip, Phys. E, 2012, 44, 1692–1696 CrossRef PubMed.
- A. Vijayalakshmi, P. R. Kumar, S. S. Priyadarsini and C. Meenaxshi, J. Chem., 2013, 1–9 CrossRef PubMed.
- P. Batra and A. K. Sharma, Biotech., 2013, 3, 439–459 Search PubMed.
- J. Kasthuri, S. Veerapandian and N. Rajendiran, Colloids Surf., B, 2009, 68, 55–60 CrossRef CAS PubMed.
- Q. Guo, Q. Guo, J. Yuan and J. Zeng, Colloids Surf., A, 2014, 441, 127–132 CrossRef CAS PubMed.
- D. K. Das, A. Chakraborty, S. Bhattacharjee and S. Dey, J. Exp. Nanosci., 2013, 8, 649–655 CrossRef CAS PubMed.
- B. K. Jena, B. K. Mishra and S. Bohidar, J. Phys. Chem. C, 2009, 113, 14753–14758 CAS.
- J. Cao, W. Chen, Y. Zhang, Y. Zhang and X. Zhao, Food Sci. Technol. Res., 2010, 16, 395–402 CrossRef CAS.
- S. Balasubramanian and R. L. Eckert, Toxicol. Appl. Pharmacol., 2007, 224, 214–219 CrossRef CAS PubMed.
- H. Wei, L. Tye, E. Bresnick and D. F. Birt, Cancer Res., 1990, 50, 499–502 CAS.
- S. Shukla and S. Gupta, Free Radical Biol. Med., 2008, 44, 1833–1845 CrossRef CAS PubMed.
- J. Fang, C. Xia, Z. Cao, J. Z. Zheng, E. Reed and B. H. Jiang, FASEB J., 2005, 19, 343–353 CrossRef PubMed.
- J. Fang, Q. Zhou, L. Z. Liu, C. Xia, X. Hu, X. Shi and B. H. Jiang, Carcinogenesis, 2007, 28, 858–864 CrossRef CAS PubMed.
- H. Shao, K. Jing, E. Mahmoud, H. Huang, X. Fang and C. Yu, Mol. Cancer Ther., 2013, 12, 2640–2650 CrossRef CAS PubMed.
- Y. Zhu, Y. Mao, H. Chen, Y. Lin, Z. Hu, J. Wu, X. Xu, X. Xu, J. Qin and L. Xie, Cancer Cell Int., 2013, 13, 1–7 CrossRef PubMed.
- X. Cao, B. Liu, W. Cao, W. Zhang, F. Zhang, H. Zhao, R. Meng, L. Zhang, R. Niu, X. Hao and B. Zhang, Chin. J. Cancer Res., 2013, 25, 212–222 Search PubMed.
- S. Das, J. Das, A. Samadder, N. Boujedaini and A. R. K. Bukhsh, Exp. Biol. Med., 2012, 237, 1433–1448 CrossRef CAS PubMed.
- S. Das, J. Das, A. Samadder, A. Paul and A. R. K. Bukhsh, Food Chem. Toxicol., 2013, 62, 670–680 CrossRef CAS PubMed.
- S. Kondath, S. R. Bhuvanasree, A. Rajaram and R. Rajaram, Chem.-Biol. Interact., 2014, 224, 78–88 CrossRef CAS PubMed.
- B. Li, D. H. Robinson and D. F. Birt, J. Pharmacol. Sci., 1997, 86, 721–725 CrossRef CAS PubMed.
- K. Sindhu, A. Rajaram, K. J. Sreeram and R. Rajaram, RSC Adv., 2014, 4, 1808–1818 Search PubMed.
- B. K. Pong, H. I. Elim, J. X. Chong, W. Ji, B. L. Trout and J. Y. Lee, J. Phys. Chem. C, 2007, 111, 6281–6287 CAS.
- A. Rai, A. Singh, A. Ahmad and M. Sastry, Langmuir, 2006, 22, 736–741 CrossRef CAS PubMed.
- D. S. D. Santos, J. Ramon, A. A. Puebla, O. N. Oliveira and R. F. Aroca, J. Mater. Chem., 2005, 15, 3045–3049 RSC.
- G. Mariappan, N. Sundaraganesan and S. Manoharan, Spectrochim. Acta, Part A, 2012, 95, 86–99 CrossRef CAS PubMed.
- B. P. Pawlęga, L. E. Misiak, B. Zarzyka, R. P. A. Gawron and W. I. Gruszecki, Biochim. Biophys. Acta, 2013, 1828, 518–527 CrossRef PubMed.
- R. Pal, S. Panigrahi, D. Bhattacharyya and A. S. Chakraborti, J. Mol. Struct., 2013, 1046, 153–163 CrossRef CAS PubMed.
- R. Shukla, V. Bansal, M. Chaudhary, A. Basu, R. R. Bhonde and M. Sastry, Langmuir, 2005, 21, 10644–10654 CrossRef CAS PubMed.
- K. Song, P. Xu, Y. Meng, F. Geng, J. Li, Z. Li, J. Xing, J. Chen and B. Kong, Int. J. Oncol., 2013, 42, 597–608 CAS.
- P. Arunkumar, H. Vedagiri and K. Premkumar, J. Nanopart. Res., 2013, 15, 1–8 CrossRef.
- J. Daduang, A. Palasap, S. Daduang, P. Boonsiri, P. Suwannalert and T. Limpaiboon, Asian Pac. J. Cancer Prev., 2015, 16, 169–174 CrossRef.
- S. Varun and S. Sellappa, Int. J. Pharm. Pharm. Sci., 2014, 6, 528–531 Search PubMed.
- G. Zhang, V. Gurtu, S. R. Kain and G. Yan, BioTechniques, 1997, 23, 525–531 CAS.
- A. Thirunavukkarasu, P. Durai, G. Ravi, G. Kasivelu, M. Ramar, A. Chinnasamy and S. Ganesan, Colloids Surf., B, 2014, 123, 549–556 CrossRef PubMed.
- P. Pozarowski and Z. Darzynkiewicz, Methods Mol. Biol., 2004, 281, 301–311 CAS.
- K. T. Butterworth, J. A. Coulter, S. Jain, J. Forker, S. J. McMahon, G. Schettino, K. M. Prise, F. J. Currell and D. G. Hirst, Nanotechnology, 2010, 23, 1–18 Search PubMed.
- M. Jeyaraj, R. Arun, G. Sathishkumar, D. MubarakAli, M. Rajesh, G. Sivanandhan, G. Kapildev, M. Manickavasagam, N. Thajuddin and A. Ganapathi, Mater. Res. Bull., 2014, 52, 15–24 CrossRef CAS PubMed.
- I. Ojea-Jimenez, N. G. Bastus and V. Puntes, J. Phys. Chem. C, 2011, 115, 15752–15757 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra04303d |
| This journal is © The Royal Society of Chemistry 2015 |