
Highly active sustainable ferrocenated iron oxide nanocatalysts for the decolorization of methylene blue†
Abstract
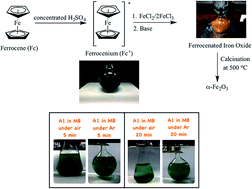
Ferrocenated iron oxide nanoparticles were successfully synthesized in basic conditions. It was unexpected to discover highly active sustainable nanocatalysts for the decolorization of methylene blue in the absence of light and hydrogen peroxide. Cyclopentadienyl radicals may be responsible for the production of active species for the decolorization of methylene blue. The nanocatalysts can be reactivated in sodium chloride solution and reused several times.
 Please wait while we load your content...
Please wait while we load your content...

