
NIS-mediated intramolecular oxidative α-functionalization of tertiary amines: transition metal-free synthesis of 1,2-dihydro-(4H)-3,1-benzoxazin-4-one derivatives†
Le Liu,
Liang Du,
Daisy Zhang-Negrerie and
Yunfei Du*
School of Pharmaceutical Science and Technology, Tianjin University, Tianjin 300072, China. E-mail: duyunfeier@tju.edu.cn; Fax: +86-22-27404031; Tel: +86-22-27404031
First published on 20th March 2015
Abstract
A novel method for direct α-functionalization of tertiary amines via NIS-mediated oxidative C–O bond formation, where NIS serves as both an oxidant and an iodination reagent, has been developed. The method provides easy access to either iodinated or non-iodinated 1,2-dihydro-(4H)-3,1-benzoxazin-4-ones, depending on the nature of the reactant, via a regioselective transition metal-free approach.
Introduction
Cross-dehydrogenative coupling (CDC) reactions, which spare pre-functionalization of the substrates, boast high synthetic efficiency and atom economy; and have, hence, attracted much attention in recent decades.1 Among the CDC reactions, selective functionalization of sp3 C–H bonds adjacent to a nitrogen atom in simple amines is of fundamental significance in organic chemistry for the synthesis of nitrogen-containing compounds.2 Classical strategies to realize such transformations have been achieved by utilizing transition-metals such as Ru,3 Fe,4 Cu,5 V,6 etc., with co-oxidant to afford an iminium cationic species, which subsequently reacts with various nucleophiles to form a C–C or C-heteroatom bond for the functionalization of the C–H bond adjacent to the nitrogen. As one of the most important branches of CDC reactions, direct C–O bond formation accomplished via selective sp3 C–H bond functionalization of tertiary amines has been intensively studied for many years.7 In 2006, a novel Pd-catalyzed highly selective acetoxylation of Boc-protected N-methylamines using IOAc as the oxidant and involving a Boc-directed C–H activation process, was developed by Yu and co-workers8 (Scheme 1, path a). In 2013, Maycock7a group reported a copper-catalyzed regioselective intramolecular α-functionalization of tertiary amines to synthesize dihydro-1,3-oxazines via C–O bond formation. Similar works have also been achieved in 2014 by Jana7b and Maiti5f employing stoichiometric Ag2O or catalytic amount of CuCl2·H2O under air, respectively (Scheme 1, path b). Lately, we reported a hypervalent iodine-mediated synthesis of 1,2-dihydro-(4H)-3,1-benzoxazin-4-one derivatives and 1,2-dihydro-(4H)-3,1-benzoxazine derivatives through intramolecular functionalization of sp3 C–H bond of diaryl amines,7c an attractive metal-free protocol to functionalize sp3 C–H bond adjacent to the nitrogen (Scheme 1, path c). Despite the significant progress made in this area in recent years,9 there is still much to be improved notably in the reaction selectivity, the expansion of substrate scope as well as the variety of the products. | ||
| Scheme 1 Direct C–O bond formation via activation of the C–H bond adjacent to nitrogen in a tertiary amine. | ||
Benzoxazin-4-one derivatives10 are an important class of heterocyclic compounds, which exhibit a wide range of biological activities including anti-inflammatory effects,11 inhibitor of human leukocyte elastase,12 HSV-1 protase,13 and have been used as potent inhibitors of chymotrypsin.14 Iodinated 1H-benzoxazin-4-(H)-one derivatives can not only be readily functionalized by various coupling reactions for further application, but also be reported as inhibitors of MEK for the treatment of a variety of proliferative disease.15 The importance of these benzoxazinones also resides in their occurrence in natural products and the application as precursors for the preparation of other heterocyclic compounds.16 Traditionally, the general method for the synthesis of benzoxazin-4-one derivatives involves the condensation between an aromatic aldehyde and an anthranilic acid in the presence of an anhydride.17 Kunai reported a straightforward access to benzoxazinones by CO2 incorporation with a zwitterion arising from nucleophilic addition of imines to arynes.18 However, efficient approaches to construct these molecules are still limited in number. On the other hand, N-halosuccinimides, aside from serving as halogenation reagents, have also been widely adopted as an oxidant to realize oxidative cyclization reactions in recent years.19 We herein disclose a novel N-iodosuccinimide (NIS)-mediated protocol to access 1,2- dihydro-(4H)-3,1-benzoxazin-4-one derivatives via α-functionalization of sp3 C–H bond adjacent to the nitrogen of tertiary amines. To our knowledge, this is an unusual reported instance to form benzoxazinones or regioseletively iodinated benzoxazinones where NIS serves both as an oxidant and an iodination reagent (Scheme 1).
Results and discussion
Initially, we postulated that treating N-methyl-N-phenylanthranilic acid 1a with NIS would probably generate an iminum intermediate which could be attacked by the neighboring carboxylic acid group to afford the aminal 2a′. However, only 15% yield of 2a′ was formed as the minor product, along with the major product of iodinated 1H-benzoxazin-4(H)-one 2a formed in 65% yield. This result suggested a regioselective iodo- aromatic substitution occurred in addition to the expected oxidative annulation.Following up with the initial result, we set out to explore the reaction conditions in hopes to enhance the chemoselectivity of the reaction, the main research interest of ours in identifying metal-free methods for the preparation of heterocycles.20 The first study was on the effect of the oxidant amount. The results, with the amount of oxidant varying between 1.0 and 3.0 equiv., showed that compound 2a was consistently the major product while 2a′, the minor product (not shown). The solvent effect study showed that acetonitrile was the most effective in comparison to all the other solvents including DCE, ethyl acetate, TFE and DMF (Table 1, entries 1–6). None of the solvents, however, showed any significant influence on the chemoselectivity of the reaction. Further studies revealed that a basic additive could dramatically improve the selectivity as well as the yield (Table 1, entries 7–10), as we were able to isolate compound 2a as the single product with a yield of 93% when Na2CO3 was applied as an additive (Table 1, entries 8). Additional bases such as K2CO3, Li2CO3 and pyridine were also examined, but no further enhancement of the yield was observed (not shown). Finally, molecular iodine was tested as a possible oxidant, yet only 10% yield of 2a was isolated (Table 1, entries 11–12). Other N-halosuccinimides, NBS and NCS, were also investigated. When NBS was used as the oxidant, neither the corresponding annulated/brominated product nor 2a′ was observed. In contrast, when NCS was employed, the reaction proceeded smoothly to give the non-halogenated 2a′ in moderate 47% yield (not shown).
|
|||||
|---|---|---|---|---|---|
| Entry | Oxidant (equiv.) | Solvent | Additive (equiv.) | Yieldb (%) of 2a′ | Yieldb (%) of 2a |
| a All reactions were carried out with 1a (0.4 mmol) and oxidant in the specific solvent (c = 0.05 M).b Isolated yield.c 15% of the starting material was recovered. ND = no desired product. | |||||
| 1c | NIS (2.0) | CH3CN | — | 15% | 67 |
| 2 | NIS (3.0) | CH3CN | — | 15% | 73 |
| 3 | NIS (3.0) | DCE | — | 20% | 68 |
| 4 | NIS (3.0) | EA | — | 17% | 70 |
| 5 | NIS (3.0) | THF | — | 23% | 65 |
| 6 | NIS (3.0) | DMF | — | 18% | 45 |
| 7 | NIS (3.0) | CH3CN | NaHCO3 (2.0) | <5 | 85 |
| 8 | NIS (3.0) | CH3CN | Na2CO3 (2.0) | — | 93 |
| 9 | NIS (3.0) | CH3CN | NaOAc (2.0) | <5 | 79 |
| 10 | NIS (3.0) | CH3CN | TEA (2.0) | Trace | Trace |
| 11 | I2 (2.0) | CH3CN | — | ND | ND |
| 12 | I2 (2.0) | CH3CN | Na2CO3 (2.0) | Trace | 10 |

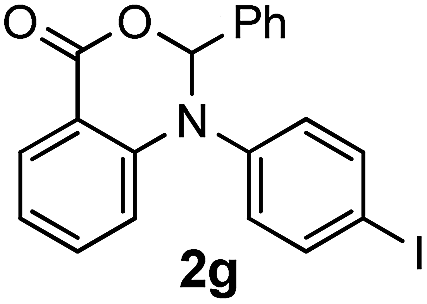
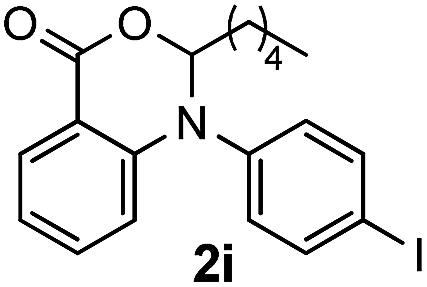
To evaluate the efficiency and generality of the newly established method, various substituted diaryl tertiary amines were examined under the optimized conditions (Table 1, entry 8). As shown in Table 2, the corresponding 1,2-dihydro-(4H)-3,1- benzoxazin-4-ones bearing a wide range of substitutions could be obtained in moderate to excellent yields. The substituent effect of R1 proved to be minimal, with well tolerated both electron-withdrawing and -donating R1 groups to give satisfactory yields (Table 2, entries 1–6). Larger alkyl groups of various sizes, such as benzyl, ethyl and hexyl were used to replace the methyl group as R3. While the yields all remained unwaveringly high (Table 2, entries 7–9), the stability of the generated product decreased dramatically in those containing a long-chained alkyl group, as 2h and 2i were found to decompose readily during purification. Drastically different from the cases of R1 and R3, the substituent effect of R2 was more pronounced. For substrates bearing one or more electron-donating groups on the B ring, diiodination occurred and diiodinated/annulated products were formed in moderate to good yields (Table 2, entries 10–12).
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yieldb (%) |
| a All reactions were carried out with 1 (0.4 mmol) in the presence of NIS (3.0 equiv.) and Na2CO3 (2.0 equiv.) in CH3CN (c = 0.05 M).b Isolated yield.c NMR yield employing mesitylene as the internal standard. | |||
| 1 | 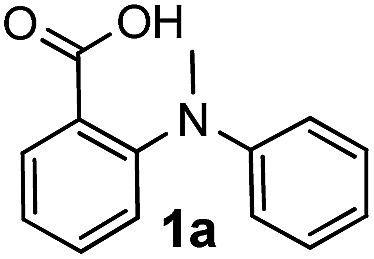 |
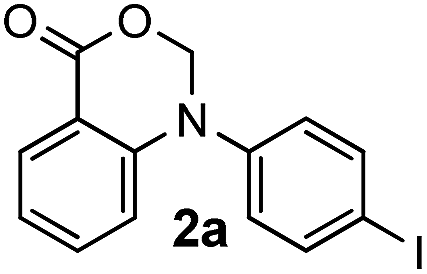 |
93 |
| 2 |  |
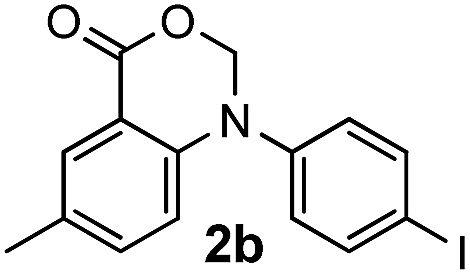 |
75 |
| 3 | 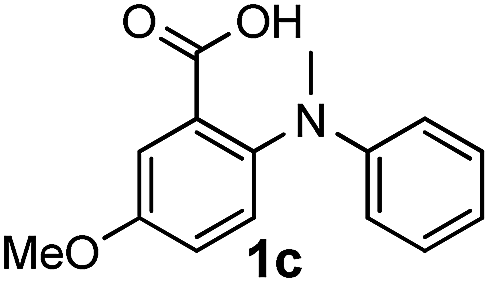 |
 |
90 |
| 4 | 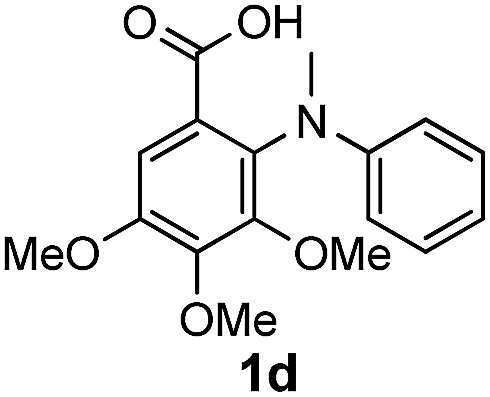 |
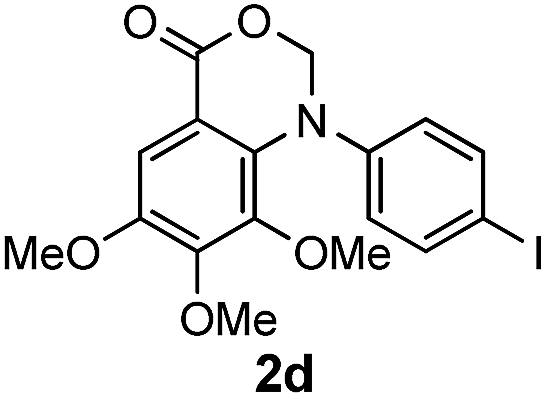 |
77 |
| 5 |  |
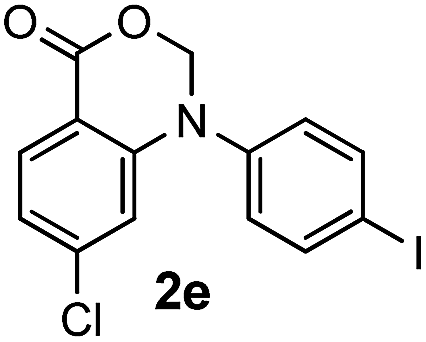 |
81 |
| 6 |  |
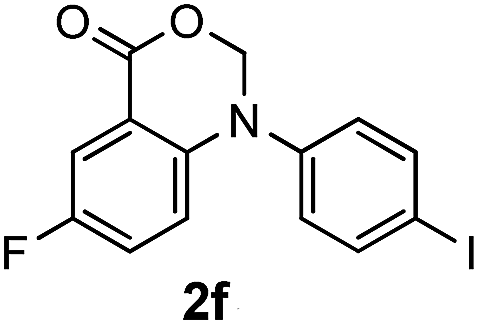 |
76 |
| 7 | 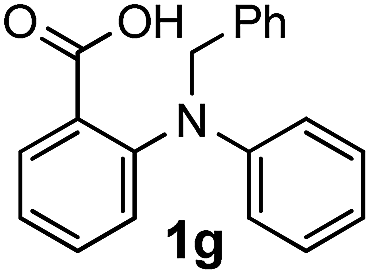 |
 |
95 |
| 8 |  |
 |
88c |
| 9 | 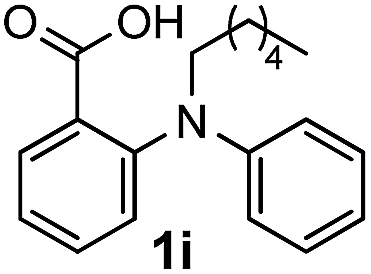 |
 |
87c |
| 10 | 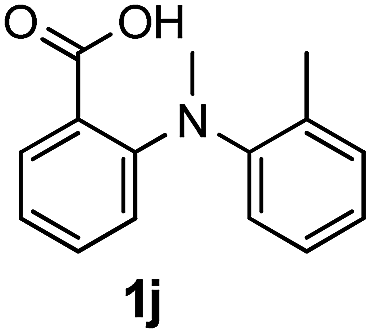 |
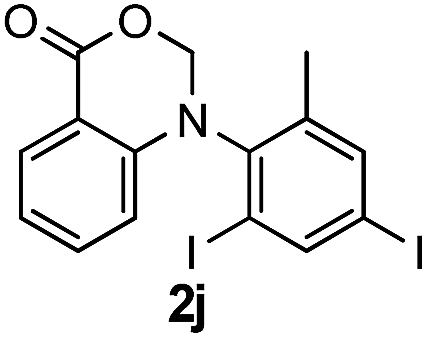 |
63 |
| 11 | 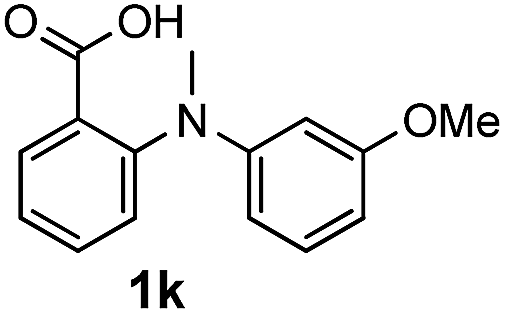 |
 |
68 |
| 12 | 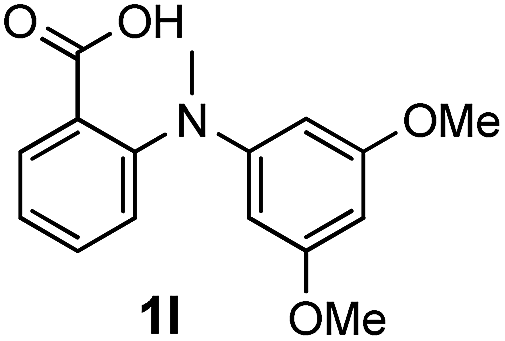 |
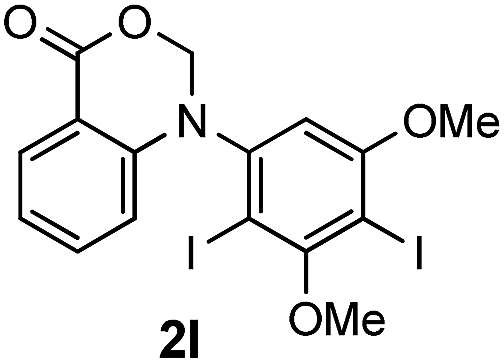 |
41 |

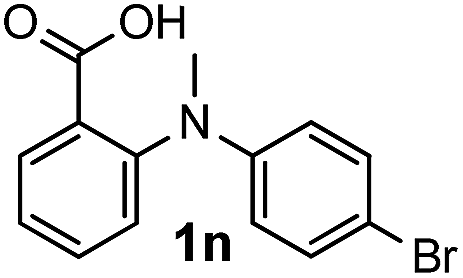
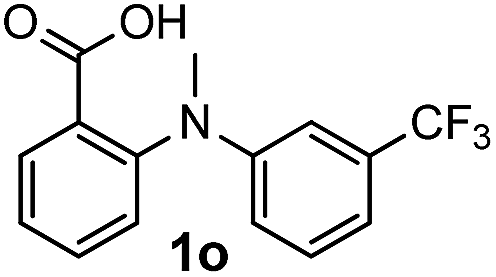
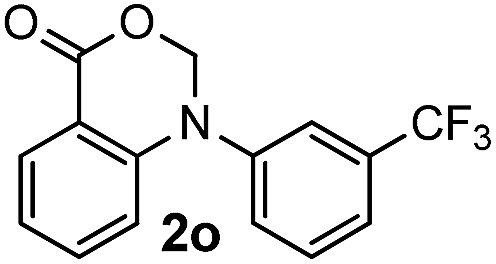
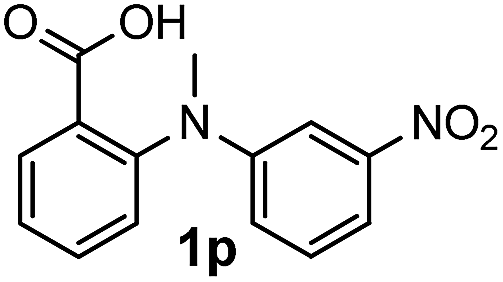
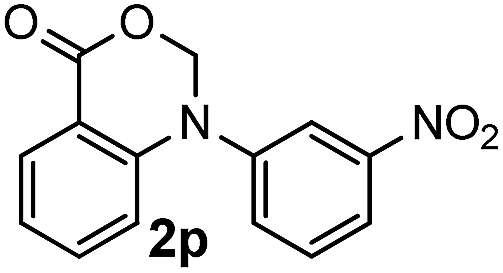
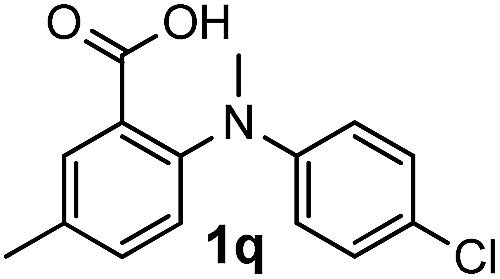
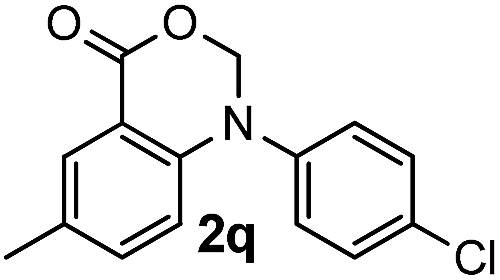
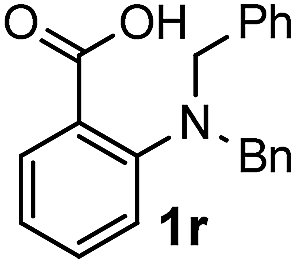
For substrates bearing an electron-withdrawing R2 group, no iodination occurred but oxidative annulation proceeded smoothly (Table 3, entries 1–5). Electron-withdrawing R2 groups such as halogens, NO2 and CF3 were all tolerated and the non-iodinated products were all obtained in great yields (Table 3, entries 1–4), including one case where there was an electron-donating –CH3 group on the A ring (Table 3, entry 5). The dibenzylic substituted tertiary amine 1r was also accessible and afforded the annulated product 2r in a relatively low yield of 43%. To our disappointment, for N-methyl amines bearing protecting groups such as Ac or Ts, no desired reaction took place (not shown).


Attempts to expand the carboxylic acid functional group to a larger range of nucleophilic moieties for the same transformation turned out to be unsuccessful. As shown in Scheme 2, subjecting 3, where the carboxylic acid in 1a was replaced by an NH–OMe amide, to the same reaction under optimized conditions, a high yield of 1,4-benzodiazepine derivative 420 was afforded instead. When the acid moiety was switched to an alcohol, no annulation took place but only para iodination which yielded amine 6 as the final product.
 | ||
| Scheme 2 Further investigation of scope of the reaction. | ||
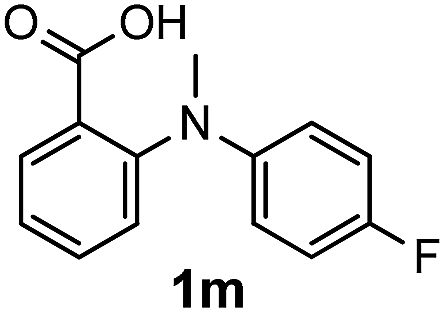
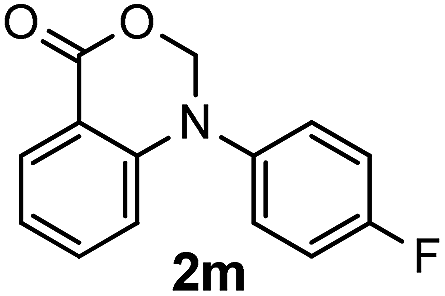
To gain more insight into the reaction mechanism, some control experiments were carried out. Treatment of the cyclized compound 2a′ with NIS in the presence of Na2CO3 under the optimized conditions led to a completely recovery of the starting material, and this suggested that iodination occurs prior to cyclization. The hypothesis was supported by the successful conversion of substrates 1m and 1n to the corresponding annulated product in 91% and 85% yields, respectively. Another control experiment was to carry out the experiment in the presence of 2 equiv. of a radical scavenger TEMPO. The unaffected high yield of the expected product excluded a radical process during the transformation.
On the basis of all the experiment results, we postulate a mechanistic pathway. Illustrated in Scheme 3, deprotonation of the acid resulted in the benzoate A, which underwent a typical electrophilic substitution reaction of aromatic ring to form C. Oxidation with the second equiv. of NIS resulted in the formation ammonium D which was subsequently transformed into the corresponding iminum intermediate E. Finally, intramolecular annulation led to the ultimate formation of product 2a. The regioselectivity of preferred para-substitution can be straightforwardly explained by the structure of the cationic B, which carries the electronic advantage over the meta-substituted counterparts and the stereo advantage over the ortho-substituted one. Finally, the electrophilic substitution mechanism for the iodination effectively explains the negative effect of an electron-withdrawing R2 group on the formation of the iodinated product, as well as the observation of multiple substitutions with an electron-donating R2 group.
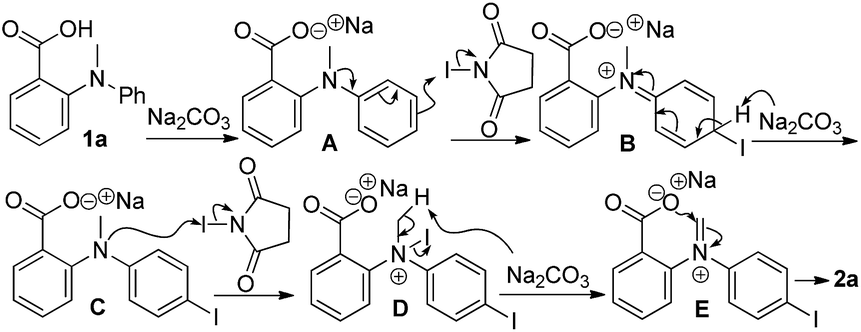 | ||
| Scheme 3 Proposed mechanism. | ||
Conclusions
In conclusion, we have developed a novel NIS-mediated intramolecular α-functionalization reaction of tertiary amines via C–O bond formation to access synthetically and biologically interesting iodinated or non-iodinated 1,2-dihydro-(4H)-3,1-benzoxazin-4-one derivatives. Besides its novelty as a CDC reaction, the one-pot procedure, mild reaction conditions, and transition metal-free feature render it a useful alternative to the existing methods for the preparation of benzoxazin-4-one derivatives.Experimental section
All reactions were carried out at room temperature under air unless otherwise noted. 1H (600 MHz or 400 MHz) and 13C (150 MHz or 100 MHz) NMR spectra were recorded at 25 °C. The chemical shifts (δ) are reported in ppm with reference to TMS (0 ppm) and coupling constants (J) are given in Hz. High resolution mass spectrometry (HRMS) was obtained on a Q-TOF micro spectro meter. Melting points were determined with a Micro Melting point apparatus without corrections. Organic solutions were concentrated by rotary evaporation below 40 °C in vacuum.General procedure for the synthesis of substrate 1
To a solution of 2-iodobenzoic acid or 2-bromobenzoic acid derivative (10 mmol) in 1,4-dioxane (c = 0.3 M) was added Cu2O (0.5 equiv.), N-methylmorpholine (1.5 equiv.), and aniline (1.5 equiv.) or its derivative. The resulting mixture was refluxed under N2 until the completion of the reaction. The mixture was filtered, and aqueous KOH solution (0.5 N, 2.5 equiv.) was added to the residue and filtered. The filtrate was extracted with DCM to remove the impurities and the pH value of the water phase was adjusted to 4 using conc. HCl. The solid generated was filtered and dried. And the crude product was used directly without further purification.To a solution of the crude product in DMF (c = 0.3 M) was slowly added NaH (2.5 equiv.) at 0 °C. And the mixture was stirred for 30 min, followed by the addition of methyl iodide (4.0 equiv.) or the corresponding haloalkane. The reaction was allowed to stir at room temperature overnight. The mixture was poured into water and extracted with EA (3 × 80 mL). The organic phase was dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography on silica gel to give the corresponding esters.
To a solution of the ester in MeOH/H2O (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, c = 0.6 M) was added KOH (2.0 equiv.), and the resulting mixture was allowed to heat to reflux. Upon completion monitored by TLC, the mixture was concentrated and poured into water. The solution was acidified with conc. HCl, and the solid was filtered to give the desired product. If no precipitation, the reaction mixture was extracted with EA (3 × 60 mL). The organic phase was dried over anhydrous Na2SO4, filtered, concentrated and purified by flash column chromatography on silica gel to give the product.
:1, c = 0.6 M) was added KOH (2.0 equiv.), and the resulting mixture was allowed to heat to reflux. Upon completion monitored by TLC, the mixture was concentrated and poured into water. The solution was acidified with conc. HCl, and the solid was filtered to give the desired product. If no precipitation, the reaction mixture was extracted with EA (3 × 60 mL). The organic phase was dried over anhydrous Na2SO4, filtered, concentrated and purified by flash column chromatography on silica gel to give the product.
Known substrates 1a,22 le,7c 1f,7c 1g,23 1h,21 1k,20 1m–q,7c 1r,7c 320 and 57c were prepared according to the literature procedures.
General procedure for the synthesis of compounds 2, 4 and 6
To a solution of N-alkyl-N-aryl anthranilic acids 1 (0.4 mmol) in CH3CN (8 mL) was added NIS (1.2 mmol) followed by the addition of Na2CO3 (0.8 mmol) at ambient temperature. The reaction mixture was maintained at room temperature and monitored by TLC. Upon completion, the mixture was poured into sat. aqueous solution of Na2S2O3 (30 mL) and extracted by ethyl acetate (3 × 50 mL). The combined organic phase was washed with brine (1 × 80 mL). Dried over anhydrous Na2SO4, evaporation of the solvent under reduced pressure and purification of the crude residue by flash column chromatography on silica gel (EA/PE with 5‰TEA) afforded the desired products. For the synthesis of compounds 2m–r, 2.0 equiv. of NIS was used following the similar procedure mentioned above.Acknowledgements
We acknowledge the National Science Foundation of China (#21472136) and the National Basic Research Project (2014CB932201) for financial support.Notes and references
- For selected examples, see: (a) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (b) W. Yoo and C. Li, Top. Curr. Chem., 2010, 292, 281 CrossRef; (c) Z. Shi and F. Glorious, Angew. Chem., Int. Ed., 2012, 51, 9220 CrossRef CAS PubMed; (d) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC; (e) R. D. Baxter, D. Sale, M. K. Engle and J. Yu, J. Am. Chem. Soc., 2012, 134, 4600 CrossRef CAS PubMed; (f) A. M. R. Smith and K. K. Hii, Chem. Rev., 2011, 111, 1637 CrossRef CAS PubMed; (g) C. Sun, B. Li and Z. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (h) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (i) A. J. Nell, J. P. Hehn, P. F. Tripet and F. D. Toste, J. Am. Chem. Soc., 2013, 133, 14044 CrossRef PubMed; (j) J. L. Jeffrey and R. Sarpong, Chem. Sci., 2013, 4, 4092 RSC.
- For selected examples, see: (a) S. Doye, Angew. Chem., Int. Ed., 2001, 40, 3351 CrossRef CAS; (b) N. Chatani, T. Asaumi, T. Ikeda, S. Yorimitsu, Y. Ishii, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 2000, 122, 12882 CrossRef CAS; (c) Z. Li and C. Li, J. Am. Chem. Soc., 2004, 126, 11810 CrossRef CAS PubMed; (d) Y. Takehiko, A. Yoshimasa and N. Hiroto, J. Am. Chem. Soc., 2005, 127, 11610 CrossRef PubMed; (e) J. Xie, H. Li, J. Zhou, Y. Cheng and C. Zhu, Angew. Chem., Int. Ed., 2012, 51, 1252 CrossRef CAS PubMed; (f) Y. Ma, G. Zhang, J. Zhang, D. Yang and R. Wang, Org. Lett., 2014, 16, 5358 CrossRef CAS PubMed; (g) H. Ueda, K. Yoshida and H. Tokuyama, Org. Lett., 2014, 16, 4194 CrossRef CAS PubMed.
- For selected examples, see: (a) D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2012, 134, 8094 CrossRef CAS PubMed; (b) M. Rueping, C. Vila, R. M. Koenigs, K. Poscharny and D. C. Fabry, Chem. Commun., 2011, 47, 2360 RSC; (c) D. B. Freeman, L. Furst, A. G. Condie and C. R. J. Stephenson, Org. Lett., 2012, 14, 94 CrossRef CAS PubMed; (d) J. Xuan, Y. Cheng, J. An, L. Lu, X. Zhang and W. Xiao, Chem. Commun., 2011, 47, 8337 RSC; (e) M. Rueping, C. Vila, R. M. Koenigs, K. Poscharny and D. C. Fabry, Chem. Commun., 2011, 47, 2360 RSC; (f) S. Kumar, P. Kumar and S. L. Jain, RSC Adv., 2013, 3, 24013 RSC.
- For selected examples, see: (a) W. Han and A. R. Ofial, Chem. Commun., 2009, 45, 5024 RSC; (b) H. Li, Z. He, X. Guo, W. Li, X. Zhao and Z. Li, Org. Lett., 2009, 11, 4176 CrossRef CAS PubMed; (c) P. Liu, Y. Liu, E. L. Wong, S. Xiang and C. Che, Chem. Sci., 2011, 2, 2187 RSC; (d) M. Brzozowski, J. A. Forni, G. P. Savage and A. Polyzos, Chem. Commun., 2015, 51, 334 RSC; (e) C. M. R. Volla and P. Vogel, Org. Lett., 2009, 11, 1701 CrossRef CAS PubMed; (f) W. Han, P. Mayer, A. and R. Ofial, Adv. Synth. Catal., 2010, 352, 1667 CrossRef CAS.
- (a) For selected examples, see: ; (b) S. A. Girard, T. Knuauber and C. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed; (c) Z. Li and C. Li, J. Am. Chem. Soc., 2005, 127, 3672 CrossRef CAS PubMed; (d) G. Zhang, Y. Zhang and R. Wang, Angew. Chem., Int. Ed., 2011, 50, 10429 CrossRef CAS PubMed; (e) W. R. Evans, J. R. Zbieg, S. Zhu, W. Li and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 16074 CrossRef PubMed; (f) A. Modak, U. Dutta, R. Kancherla, S. Maity, M. Bhadra, S. M. Mobin and D. Maiti, Org. Lett., 2014, 16, 2602 CrossRef CAS PubMed; (g) O. Basle and C. Li, Green Chem., 2008, 10, 249 RSC; (h) Y. Yan, Y. Yuan and N. Jiao, Org. Chem. Front., 2014, 1, 1176 RSC.
- For selected examples, see: (a) A. Sud, D. Sureshkumar and M. Klussmann, Chem. Commun., 2009, 45, 3169 RSC; (b) L. Rout and T. Punniyamurthy, Adv. Synth. Catal., 2005, 347, 1958 CrossRef CAS; (c) C. Zhu, J. Xia and C. Chen, Tetrahedron Lett., 2014, 55, 232 CrossRef CAS PubMed; (d) S. Singhal, S. L. Jain and B. Sain, Chem. Commun., 2009, 45, 2371 RSC.
- For selected examples, see: (a) M. L. Deb, S. S. Dey, I. Bento, M. T. Barros and C. D. Maycock, Angew. Chem., Int. Ed., 2013, 52, 9791 CrossRef CAS PubMed; (b) S. Mahato, S. Haldar and C. K. Jana, Chem. Commun., 2014, 50, 332 RSC; (c) N. Zhang, R. Cheng, D. Zhang-Negrerie, Y. Du and K. Zhao, J. Org. Chem., 2014, 79, 10581 CrossRef CAS PubMed; (d) M. O. Ratnikov and M. P. Doyle, J. Am. Chem. Soc., 2013, 135, 1549 CrossRef CAS PubMed; (e) M. O. Ratnikov, X. Xu and M. P. Doyle, J. Am. Chem. Soc., 2013, 135, 9475 CrossRef CAS PubMed; (f) S. I. Murahashi, T. Naota, N. Miyaguchi and T. Nakato, Tetrahedron Lett., 1992, 33, 6991 CrossRef CAS; (g) S. I. Murahashi, T. Naota and K. Yonemura, J. Am. Chem. Soc., 1988, 110, 8256 CrossRef CAS.
- D. Wang, X. Hao, D. Wu and J. Yu, Org. Lett., 2006, 8, 3387 CrossRef CAS PubMed.
- For selected examples on metal-free CDC reactions of tertiary amines, see: (a) Y. Pan, C. W. Kee, L. Chen and C. Tan, Green Chem., 2011, 13, 2682 RSC; (b) W. Huang, C. Ni, Y. Zhao and J. Hu, New J. Chem., 2013, 37, 1684 RSC; (c) T. Nobuta, A. Fujiya, T. Yamaguchi, N. Tada, T. Miura and A. Itoh, RSC Adv., 2013, 2, 10189 RSC; (d) D. P. Hari and B. König, Org. Lett., 2011, 13, 3852 CrossRef CAS PubMed; (e) L. Liu, Z. Wang, X. Fu and C. Yan, Org. Lett., 2012, 14, 5692 CrossRef CAS PubMed; (f) A. Tanoue, W. Yoo and S. Kobayashi, Org. Lett., 2014, 16, 2346 CrossRef CAS PubMed; (g) J. Dhineshkumar, M. Lamani, K. Alagiri and K. R. Prabhu, Org. Lett., 2013, 15, 3852 CrossRef PubMed.
- For selected examples, see: (a) M. Alajarin, A. Vidal, M. M. Ortina and D. Bautista, Synthesis, 2005, 2426 CrossRef CAS PubMed; (b) C. Parkanyi, H. L. Yuan, B. H. E. Stromberg and A. Evenzahav, J. Heterocycl. Chem., 1992, 29, 749 CrossRef CAS; (c) L. A. Errede, J. J. MeBrady and H. T. Oien, J. Org. Chem., 1977, 42, 656 CrossRef CAS; (d) P. Wiklund and J. Bergman, Org. Biomol. Chem., 2003, 1, 367 RSC; (e) N. J. Hrib, J. G. Kurcak, D. E. Bregna, K. L. Burgher, H. B. Hartman, S. Kafka, L. L. Kerman, S. Kongsamut, J. E. Roehr, M. R. Szewczak, A. T. Wood-Ketteberger and R. Corbett, J. Med. Chem., 1996, 39, 4044 CrossRef CAS PubMed.
- (a) C. Wu, T. Wang, W. Wang, P. W. Hsieh and Y. Wu, Eur. J. Pharmacol., 2005, 527, 37 CrossRef CAS PubMed; (b) P. W. Hesieh, T. L. Hwang, C. C. Wu, F. Chang, T. Wang and Y. Wu, Bioorg. Med. Chem., 2005, 15, 2786 CrossRef PubMed.
- (a) R. L. Stein, A. M. Strimpler, B. R. Viscarello, R. A. Wildonger, R. C. Mauger and D. A. Trainor, Biochemistry, 1987, 26, 4126 CrossRef CAS; (b) A. Krantz, R. W. Spencer, T. F. Tam, T. J. Liak, L. J. Copp, E. M. Thomas and S. P. Rafferty, J. Med. Chem., 1990, 33, 464 CrossRef CAS.
- R. L. Harvest, M. J. Parratt, C. M. Debouck, J. G. Gorniak, L. J. Jennings, H. T. Serafinowska and J. E. Strickler, Bioorg. Med. Chem. Lett., 1996, 6, 2463 CrossRef.
- (a) T. Teshima, J. C. Griffin and J. C. Powers, J. Biol. Chem., 1982, 257, 5085 CAS; (b) L. Hedstrom, A. R. Moorman, J. Dobbs and R. H. Abeles, Biochemistry, 1984, 23, 1753 CrossRef CAS.
- C. Biwersi, H. Tecle and J. S. Warmus, US Pat., US7001905B2, 2006.
- (a) M. Bouillant, J. Favre-Bonvin and P. Ricci, Tetrahedron Lett., 1983, 24, 51 CrossRef CAS; (b) G. J. Niemann, J. Liem, A. Vander Kerk-Van Hoff and W. M. A. Neissen, Photochemistry, 1992, 31, 3761 CrossRef CAS.
- (a) F. Nikpour, D. Sheikh and M. Saraji, Chem. Lett., 2007, 36, 1074 CrossRef CAS; (b) D. Azarifar and D. Sheikh, Heteroat. Chem., 2011, 22, 106 CrossRef CAS.
- H. Yoshida, H. Fukushima, J. Ohshita and A. Kunai, J. Am. Chem. Soc., 2006, 128, 11040 CrossRef CAS PubMed.
- For selected examples, see: (a) B. Godoi, R. F. Schumacher and G. Zeni, Chem. Rev., 2011, 111, 2937 CrossRef CAS PubMed; (b) A. Palisse and S. F. Kirsch, Org. Biomol. Chem., 2012, 10, 8041 RSC; (c) S. R. Bathula, M. P. Reddy, K. K. D. R. Viswanaham, P. Sathyanarayana and M. S. Reddy, Eur. J. Org. Chem., 2013, 4552 CrossRef CAS; (d) X. Wang, D. Xu, C. Miao, Q. Zhang and W. Sun, Org. Biomol. Chem., 2014, 12, 3018 Search PubMed; (e) L. Zhang, Q. Zeng, A. Mao, Z. Wu, T. Luo, Y. Xiao and J. Zhang, Org. Biomol. Chem., 2014, 12, 8942 RSC; (f) Y. Hu, R. Yi, C. Wang, X. Xin, F. Wu and B. Wan, J. Org. Chem., 2014, 79, 3052 CrossRef CAS PubMed; (g) Y. Yan, Y. Zhang, Z. Zha and Z. Wang, Org. Lett., 2013, 15, 2274 CrossRef CAS PubMed.
- For selected examples of our previous work on metal-free methods, see: (a) Y. Du, R. Liu, G. Linn and K. Zhao, Org. Lett., 2006, 8, 5919 CrossRef CAS PubMed; (b) W. Yu, Y. Zhang, S. Wang and K. Zhao, Org. Lett., 2009, 11, 2417 CrossRef CAS PubMed; (c) X. Li, Y. Du, Z. Liang, X. Li, Y. Pan and K. Zhao, Org. Lett., 2009, 11, 2643 CrossRef CAS PubMed; (d) J. Wang, Y. Yuan, R. Xiong, D. Zhang-Negrerie, Y. Du and K. Zhao, Org. Lett., 2012, 14, 2210 CrossRef CAS PubMed; (e) L. Liu, H. Lu, H. Wang, C. Yang, X. Zhang, D. Zhang-Negrerie, Y. Du and K. Zhao, Org. Lett., 2013, 15, 2906 CrossRef CAS PubMed; (f) S. Shang, D. Zhang-Negrerie, Y. Du and K. Zhao, Angew. Chem., Int. Ed., 2014, 53, 6252 Search PubMed; (g) Y. Zheng, X. Li, R. Ren, D. Zhang-Negrerie, Y. Du and K. Zhao, J. Org. Chem., 2012, 77, 10353 CrossRef CAS PubMed.
- For our previous work on synthesis of 1,4-benzodiazepine derivatives, see: X. Li, Y. Liu, X. Zhang, D. Zhang-Negrerie, Y. Du and K. Zhao, J. Org. Chem., 2014, 79, 955 CrossRef CAS PubMed.
- (a) Z. Zheng, L. Dian, Y. Yuan, D. Zhang-Negrerie, Y. Du and K. Zhao, J. Org. Chem., 2014, 79, 7451 CrossRef CAS PubMed; (b) J. Mortier, A. Castant and M. Belaud-Rotureau, US Pat., US2012316337A1, 2012.
- E. M. Davis, T. N. Nanninga, H. I. Tjiong and D. D. Winkle, Org. Process Res. Dev., 2005, 9, 843 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra for all new compounds, See DOI: 10.1039/c5ra03882k |
| This journal is © The Royal Society of Chemistry 2015 |