
Ni/Mn ratio and morphology-dependent crystallographic facet structure and electrochemical properties of the high-voltage spinel LiNi0.5Mn1.5O4 cathode material†
Abstract
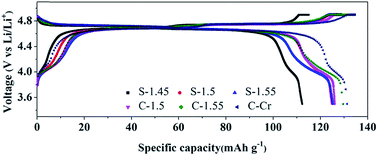
The LiNi0.5Mn1.5O4 (LNMO) spinel is an attractive cathode material for next generation lithium-ion batteries as it offers a high power capability with a discharge voltage of 4.7 V and a theoretical capacity of 147 mA h g−1. In this paper, porous LNMO microspheres/cubes, which are constructed with nanometer-sized primary particles with different Ni/Mn ratios, have been synthesized by a facile method that involves the use of MnCO3 microspheres/cubes as the self-supporting template. The effects of the morphology of the MnCO3 and the Ni/Mn ratio on the physicochemical and electrochemical properties of the as-synthesized LNMO materials were investigated in detail. Scanning electron microscopy (SEM) observation shows that the morphology of the porous MnCO3 has an important effect on the morphology and degree of dispersion of the obtained LNMO spinels, so as to the electrochemical performance. XPS and XRD results show that the Ni/Mn ratio has a significant impact on the Mn3+ content, phase purity (rock-salt phase) and crystallographic facet orientations of the LNMO-based cathode materials. In particular, the Mn3+ content, rock-salt phase and high-active (111) facet in the LNMO spinels were found to be adjusted by the Ni/Mn ratio. With the presence of a reasonable Mn3+ content, high-active facet, the absence of the impurity phase (rock-salt phase) as well as the large cationic disorder in the Cr-doping LNMO spinels, the rate performance and capacity retention of the product could be significantly improved. All these findings show the important roles of the synergic effects of the morphology and the composition on the improvement of the electrochemical performance of LNMO-based cathode materials.
 Please wait while we load your content...
Please wait while we load your content...

