
MnII and CuII complexes with arylhydrazones of active methylene compounds as effective heterogeneous catalysts for solvent- and additive-free microwave-assisted peroxidative oxidation of alcohols†
Kamran T. Mahmudov*ab,
Manas Sutradhara,
Luísa M. D. R. S. Martins*ac,
M. Fátima C. Guedes da Silva*a,
Alice Riberaad,
Ana V. M. Nunese,
Shahnaz I. Gahramanovaf,
Fabio Marchettid and
Armando J. L. Pombeiro*a
aCentro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049–001 Lisbon, Portugal. E-mail: kamran_chem@yahoo.com; kamran_chem@mail.ru; fatima.guedes@tecnico.ulisboa.pt; lmartins@deq.isel.ipl.pt; pombeiro@tecnico.ulisboa.pt
bBaku State University, Department of Chemistry, Z. Xalilov Str. 23, Az 1148 Baku, Azerbaijan
cChemical Engineering Department, ISEL, R. Conselheiro Emídio Navarro, 1959-007 Lisboa, Portugal
dSchool of Pharmacy, University of Camerino, Via S. Agostino 1, 62032 Camerino, Italy
eRequimte/CQFB, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Campus de Caparica, Caparica 2829-516, Portugal
fInstitute of Catalysis and Inorganic Chemistry named after acad. M.F. Nagiyev, H. Cavid 113, Az 1143 Baku, Azerbaijan
First published on 5th March 2015
Abstract
A one-pot template reaction of sodium 2-(2-(dicyanomethylene)hydrazinyl)benzenesulfonate (NaHL1) with water and manganese(II) acetate tetrahydrate led to the mononuclear complex [Mn(H2O)6](HL1a)2·4H2O (1), where (HL1a)− = 2-(SO3−)C6H4(NH)N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(C
C(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) (CONH2) is the carboxamide species derived from nucleophilic attack of water on a cyano group of (HL1)−. The copper tetramer [Cu4(H2O)10(1κN:κ2O:κO,2κN:κO-L2)2]·2H2O (2) was obtained from reaction of Cu(NO3)2·2.5H2O with sodium 5-(2-(4,4-dimethyl-2,6-dioxocyclohexylidene)hydrazinyl)-4-hydroxybenzene-1,3-disulfonate (Na2H2L2). Both complexes were characterized by elemental analysis, IR spectroscopy, ESI-MS and single crystal X-ray diffraction. They exhibit a high catalytic activity for the solvent- and additive-free microwave (MW) assisted oxidation of primary and secondary alcohols with tert-butylhydroperoxide, leading to yields of the oxidized products up to 85.5% and TOFs up to 1.90 × 103 h−1 after 1 h under low power (5–10 W) MW irradiation. Moreover, the heterogeneous catalysts are easily recovered and reused, at least for three consecutive cycles, maintaining 89% of the initial activity and a high selectivity.
N) (CONH2) is the carboxamide species derived from nucleophilic attack of water on a cyano group of (HL1)−. The copper tetramer [Cu4(H2O)10(1κN:κ2O:κO,2κN:κO-L2)2]·2H2O (2) was obtained from reaction of Cu(NO3)2·2.5H2O with sodium 5-(2-(4,4-dimethyl-2,6-dioxocyclohexylidene)hydrazinyl)-4-hydroxybenzene-1,3-disulfonate (Na2H2L2). Both complexes were characterized by elemental analysis, IR spectroscopy, ESI-MS and single crystal X-ray diffraction. They exhibit a high catalytic activity for the solvent- and additive-free microwave (MW) assisted oxidation of primary and secondary alcohols with tert-butylhydroperoxide, leading to yields of the oxidized products up to 85.5% and TOFs up to 1.90 × 103 h−1 after 1 h under low power (5–10 W) MW irradiation. Moreover, the heterogeneous catalysts are easily recovered and reused, at least for three consecutive cycles, maintaining 89% of the initial activity and a high selectivity.
1. Introduction
The development of efficient and selective catalysts for the oxidation of alcohols into their corresponding carbonyl compounds is a fundamental issue in organic synthesis, as well as in the chemical industry.1,2 Carbonyl compounds, such as aldehydes and ketones, are precursors for the synthesis of many drugs, ligands, vitamins, etc., and important intermediates for many complex syntheses.1–3 A good number of studies has been reported on homogeneous or heterogeneous catalytic oxidation of alcohols with different catalysts and oxidants.4,5 Due to several disadvantages of heterogeneous systems, namely difficult catalyst recycling, alcohol oxidation using heterogeneous catalysts is gathering increasing interest.2,4 Moreover, oxidation of alcohols with air or peroxides represents an important field of contemporary metal complex catalysis.4–6 Galactose oxidase is a copper-containing enzyme that catalyzes the two-electron oxidation of primary alcohols to aldehydes using molecular oxygen as the terminal oxidant.4 Synthetic models of galactose oxidase, i.e., copper complexes with different types of organic ligands are also known to be good catalysts in the oxidation of alcohols by molecular oxygen of air or hydrogen peroxide.4,5Recently, we have found that water-solubility of arylhydrazones of active methylene compounds (AHAMCs) and their copper(II) complexes can be increased by functionalization of ligands with hydrophilic polar groups, such as sulfo, carboxy or nitro, and they can be significantly active catalysts in the aerobic and peroxidative oxidation of alcohols or of unsaturated hydrocarbons to the corresponding organic compounds.7–11 The modification of AHAMCs ligands with several sulfonic groups can promote the solubility of the isolated complexes in polar solvents, but hamper their solubility in solvents with low polarity. Hence, these complexes could act as heterogeneous catalysts for the solvent-free and microwave-assisted oxidation of less polar bulky substituted or long chain alcohols, such as benzyl alcohol, 1-phenylethanol, hexan-1-ol, heptan-1-ol, etc. to the corresponding carbonyl compounds. The application of such an approach to the establishment of an heterogeneous catalytic system for alcohol oxidation constitutes the main aim of the current study.
Herein, we report the synthesis and characterization of highly water soluble copper(II) and manganese(II) complexes of AHAMCs, i.e., 2-(2-(dicyanomethylene)hydrazinyl)benzenesulfonate (HL1)− and 5-(2-(4,4-dimethyl-2,6-dioxocyclohexylidene)hydrazinyl)-4-hydroxybenzene-1,3 disulfonate (L2)4−, and their catalytic activity (as heterogeneous catalysts) towards solvent- and additive-free peroxidative oxidation of aromatic and of aliphatic cyclic or long chain alcohols under low power microwave irradiation.
2. Results and discussion
2.1. Synthesis and characterization of 1 and 2
Sodium salts of both ligands, NaHL1 and Na2H2L2, were synthesized by the Japp–Klingemann method7b,12 upon reaction between the aromatic diazonium salt of a substituted aniline and malononitrile or 5,5-dimethylcyclohexane-1,3-dione in water solution containing sodium hydroxide (for analytical data see ESI†). They were reported earlier by some of us.7b,12The reaction of NaHL1 with Mn(CH3COO)2·4H2O in water/acetone mixture afforded a deep red precipitate which upon recrystallization from methanol produces deep red crystals of [Mn(H2O)6](HL1a)2·4H2O (1) in 70% yield (Scheme 1). The synthesis involves an in situ ligand formation, in which one of the cyano groups of (HL1)− undergoes hydrolysis by water resulting in the formation of the carboxamide (HL1a)− = 2-(SO3−)C6H4(NH)NC(CN)(CONH2). A slow evaporation of a mixture of copper(II) nitrate hydrate, Na2H2L2 and HNO3 in water/methanol mixture furnishes greenish-black crystals of the tetranuclear complex [Cu(H2O)Cu(H2O)4(μ-L2)]2·2H2O (2) (Scheme 1).
 | ||
| Scheme 1 Synthesis and schematic representation of 1 and 2. | ||
Both complexes were characterized by elemental analysis, IR spectroscopy, ESI-MS and X-ray analysis. Elemental analysis and the ESI-MS peaks at 81.5 [Mn(H2O)6]2+, 163.0 [Mn(H2O)6]+, 267.2 (HL1a)−, and 652.3 [Cu2(H2O)6(L2)+H+] support the formulation. In the IR spectrum of 1, ν(OH) and ν(NH) vibrations are observed at 3436 and 3103 cm−1, respectively, while ν(CN) of the unreacted cyano group appears at 2227 cm−1. A new peak at 1645 cm−1 indicates that, in the solid state, 1 exhibits the carboxamide form which is also supported by X-ray data (see below). The IR spectrum of 2 displays peaks at 3307, 3217 and 3129 ν(OH), 1666 and 1639 ν(CO) and 1582 ν(CN) cm−1, which are significantly shifted in relation to those of the corresponding free ligand at 3287, 3203 and 3105 ν(OH), 2965 ν(NH), 1648 and 1622 ν(CO) and 1576 ν(CN) cm−1. No band assignable to ν(NH) was observed, indicating that (L2)4– is coordinated in the deprotonated hydrazone form (see below).
The molecular structures of complexes 1 and 2 have been established by single-crystal X-ray diffraction (Scheme 1, Fig. 1). The crystal structure of 1 consists of two anionic organic moieties, one cationic Mn centre with an almost perfect octahedral geometry (quadratic elongation of 1.000 and angle variance of 0.52°2)13 filled with water ligands, and two crystallization water molecules. The asymmetric unit of 2 contains half of the tetranuclear cluster molecule, an inversion centre being located in the middle of the macrocycle. The L2 ligands act as hexadentate 1κN:κ2O:κO,2κN:κO chelators to the copper cations that present distorted N1O4 square pyramidal (Cu1, τ5 = 0.14)14 and N1O5 octahedral environments (Cu2, quadratic elongation of 1.058 and angle variance of 591.06°2).13 Both compounds present extensive hydrogen bond interactions that lead to 3D supramolecular frameworks.
 | ||
| Fig. 1 Crystal structures of 1 and 2. Hydrogen atoms and crystallization water molecule (in 2) were omitted for clarity. Symmetry codes to generate equivalent atoms: (i) 1 − x, −y, −z (1); 1 − x, 1 − y, 1 − z (2). | ||
2.2. Catalytic activity of 1 and 2 in alcohol oxidation
Complexes 1 and 2 were tested as catalysts for the oxidation of 1-phenylethanol (model substrate), using aqueous tert-butylhydroperoxide (ButOOH, TBHP, aq. 70%, 2 eq.) as oxidizing agent, under typical conditions of 150 °C, low power (10 W) microwave (MW) irradiation, 1 h reaction time and in a solvent- and additive-free medium (Scheme 2). A good catalytic activity was observed under the above conditions, leading to yields of acetophenone up to 76.1% (1) or 85.5% (2) (Table 1, entries 12 and 10, respectively), and turnover frequencies (TOF, moles of product per mol of catalyst per hour) up to 1.83 × 10−3 (1) or 1.90 × 103 h−1 (2, i.e., 4.75 × 102 h−1 per Cu atom) for a low catalyst loading (catalyst/substrate molar ratio of 0.04%) after 1 h under MW irradiation (Table 1, entries 11 and 13, respectively). | ||
| Scheme 2 MW-assisted oxidation of 1-phenylethanol to acetophenone catalysed by 1 or 2. | ||
| Entry | Catalyst (amount, mol% vs. substrate) | Reaction time (h) | Temperature (°C) | Additive (mol% vs. substrate) | Yieldb (%) | TONc [TOF (h−1)] |
|---|---|---|---|---|---|---|
| a Reaction conditions unless stated otherwise: 2.5 mmol of 1-phenylethanol, 1–10 μmol (0.04–0.4 mol% vs. substrate) of 1 or 2, 5 mmol of TBHP (2 eq., 70% in H2O), 80–150 °C, MW irradiation (5–10 W power).b Molar yield (%) based on substrate, i.e. moles of acetophenone per 100 mol of 1-phenylethanol determined by GC.c Turnover number = number of moles of product per mol of catalyst precursor; TOF = TON per hour (values in brackets).d 5 mmol of H2O2 (30% aqueous solution) instead of TBHP.e Included for comparative purposes. | ||||||
| 1 | 1 (0.4) | 0.5 | 80 | — | 6.0 | 15 (30) |
| 2 | 1 (0.4) | 1 | 80 | — | 11.1 | 28 (28) |
| 3 | 1 (0.4) | 2 | 80 | — | 14.7 | 37 (18) |
| 4 | 2 (0.4) | 0.5 | 80 | — | 14.4 | 36 (72) |
| 5 | 2 (0.4) | 1 | 80 | — | 21.1 | 52 (52) |
| 6 | 2 (0.4) | 2 | 80 | — | 39.2 | 98 (49) |
| 7 | 1 (0.4) | 1 | 120 | — | 33.1 | 83 (83) |
| 8 | 1 (0.4) | 1 | 150 | — | 75.2 | 188 (188) |
| 9 | 2 (0.4) | 1 | 120 | — | 83.2 | 208 (208) |
| 10 | 2 (0.4) | 1 | 150 | — | 85.5 | 214 (214) |
| 11 | 1 (0.04) | 1 | 150 | — | 73.0 | 1.83 × 103 (1.83 × 103) |
| 12 | 1 (0.2) | 1 | 150 | — | 76.1 | 381 (381) |
| 13 | 2 (0.04) | 1 | 150 | — | 76.0 | 1.90 × 103 (1.90 × 103) |
| 14 | 2 (0.2) | 1 | 150 | — | 78.2 | 391 (391) |
| 15 | 1 (0.4) | 1 | 150 | K2CO3 (2.5) | 38.1 | 95 (95) |
| 16 | 1 (0.4) | 1 | 150 | HNO3 (2.5) | 34.0 | 85 (85) |
| 17 | 1 (0.4) | 1 | 150 | TEMPO (2.5) | 56.6 | 142 (142) |
| 18 | 1 (0.4) | 1 | 150 | Ph2NH (100) | 9.7 | 24 (24) |
| 19 | 1 (0.4) | 1 | 150 | CBrCl3 (100) | 33.1 | 83 (83) |
| 20 | 2 (0.4) | 1 | 150 | K2CO3 (2.5) | 19.8 | 50 (50) |
| 21 | 2 (0.4) | 1 | 150 | HNO3 (2.5) | 43.7 | 109 (109) |
| 22 | 2 (0.4) | 1 | 150 | TEMPO (2.5) | 58.1 | 145 (145) |
| 23 | 2 (0.4) | 1 | 150 | Ph2NH (100) | 5.6 | 14 (14) |
| 24 | 2 (0.4) | 1 | 150 | CBrCl3 (100) | 33.7 | 84 (84) |
| 25d | 1 (0.4) | 1 | 150 | — | 3.0 | 8 (8) |
| 26e | Cu(NO3)2 (0.4) | 1 | 150 | — | 18.2 | 46 (46) |
| 27e | Mn(CH3COO)2 (0.4) | 1 | 150 | — | 16.9 | 42 (42) |
Moreover, these heterogeneous catalysts could be easily recovered and reused, at least for three consecutive cycles, maintaining 89% of the initial activity and a rather high selectivity (see below).
Complexes 1 and 2 were also tested towards the oxidation of primary (benzyl alcohol) and other secondary alcohols, namely cyclohexanol and the linear 2- and 3-hexanol. The ketones are the only oxidation products obtained (Table 2). As expected, the alicyclic cyclohexanol is less reactive than 1-phenylethanol: in the presence of a catalytic amount of 1 or 2 (0.4 mol% vs. substrate) and in the solvent- and additive-free medium, the systems yield 56.4% and 65.5% of cyclohexanone after 1 h at 150 °C/10 W MW irradiation (Table 2, entries 1 and 2, for 1 and 2, respectively). The linear aliphatic alcohols 2-hexanol and 3-hexanol lead to similar (1) or lower (2) yields (Table 2, entries 3–6), under similar reaction conditions, as reported in other cases.5,6h–k,15 However, benzyl alcohol leads to the lowest product yield in contrast to what is usually observed,16 what can be due to the used temperature of 150 °C. In fact, it was previously found15a that temperatures up to 100 °C are preferable for the solvent-free peroxidative MW oxidation of this substrate catalyzed by copper compounds.
| Entry | Catalyst | Substrate | Product | Yieldb (%) | TONc [TOF (h−1)] |
|---|---|---|---|---|---|
| a Reaction conditions: 2.5 mmol of substrate, 5 mmol of TBHP (aq. 70%), 10 μmol (0.4 mol% vs. substrate) of 1 or 2, 150 °C, 1 h of microwave irradiation (10 W).b Molar yield (%) based on substrate, i.e. moles of product per 100 mol of substrate determined by GC.c Turnover number = number of moles of product per mol of metal catalyst; TOF = TON per hour (values in brackets). | |||||
| 1 | 1 | Cyclohexanol | Cyclohexanone | 56.4 | 141 (141) |
| 2 | 2 | 65.5 | 164 (164) | ||
| 3 | 1 | 2-Hexanol | 2-Hexanone | 60.3 | 151 (151) |
| 4 | 2 | 52.4 | 131 (131) | ||
| 5 | 1 | 3-Hexanol | 3-Hexanone | 55.5 | 139 (139) |
| 6 | 2 | 37.1 | 93 (93) | ||
| 7 | 1 | Benzylalcohol | Benzaldehyde | 24.0 | 60 (60) |
| 8 | 2 | 19.1 | 48 (48) | ||
While the heterogeneous copper 2/MW/TBHP oxidation system of the present study yields ketone products in the range of other previously reported copper homogeneous systems,5,6h–k the heterogeneous Mn 1/MW/TBHP system is much more efficient than other MW/TBHP homogeneous systems with dinuclear Mn(II) complexes with Schiff bases15b in the oxidation of secondary alcohols, since our system operates effectively under additive-free conditions and requires much lower loads of catalyst precursor.
The activity of 1 or 2 in the oxidation of benzyl alcohol is rather modest since only moderated 19% (1) or 24% (2) yields of benzaldehyde were obtained under the same reaction conditions (Table 2, entries 7 and 8, for 1 and 2, respectively). The silica-supported manganese dioxide previously applied16 for the oxidation of benzyl alcohol under MW solvent-free conditions, yielded 88% of benzaldehyde after 20 s irradiation. However, the MW power and the temperature were not indicated and this system required a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar excess of MnO2 relatively to the substrate.
:1 molar excess of MnO2 relatively to the substrate.
The influences of various reaction parameters, such as the amounts of catalyst and oxidant, type of oxidant, time, temperature and presence of additives, were investigated for the most active substrate (1-phenylethanol), and the results are summarized in Table 1 and Fig. 2–4.
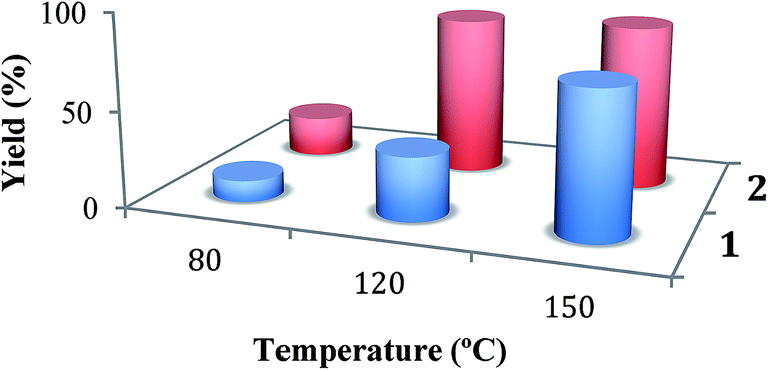 | ||
| Fig. 2 Influence of the temperature on the yield of acetophenone in the MW-assisted peroxidative oxidation of 1-phenylethanol catalysed by 1 or 2. | ||
The MW-assisted alcohol oxidation depends strongly on the temperature (Fig. 2). The overall temperature coefficient of the oxidations is ca. 6.8 for 1 or 4.1 for 2 (temperature range 80–150 °C). For example, a high yield of 85.5% of acetophenone is achieved after 1 h at 150 °C (5 W) for the copper system 2 without any additive (Table 1, entry 10), which is much higher than the 21.1% yield obtained for the same reaction time but at 80 °C (Table 1, entry 5). Furthermore, at this temperature, only 39.2% (2) or 14.7% (1) of acetophenone is obtained for the extended reaction time of 2 h (Table 1, entries 6 and 3, respectively).
The influence of the amount of 1 and 2 on the yield and TON (or TOF) is shown in Fig. 3 (entries 8 and 10–14, Table 1). While an amount increase of 1 does not appear to have a significant effect on the acetophenone yield, the increase of 2 results in a yield enhancement, e.g. from 76.0% to 85.5% upon changing the amount of catalyst from 1 μmol (0.04% vs. substrate) to 10 μmol (0.4 mol% vs. substrate). As expected, the increase of the catalysts amounts results in a corresponding TON lowering for both 1 and 2 (Fig. 3).
 | ||
| Fig. 3 Effect of the amount of catalysts 1 and 2 on the yield (solid lines) and turnover number (TON) (dashed lines) for the oxidation of 1-phenylethanol to acetophenone. | ||
For 1, experiments with the cheaper and environmentally friendly hydrogen peroxide (30% aqueous solution) as oxidant are not effective, as attested by the marked yield lowering, e.g., from 85.5% to 3.0% (entries 10 and 25, Table 1), in accord with the expected decomposition of H2O2 under the used reaction conditions (150 °C).
The influence of 2,2,6,6-tetramethylpiperidyl-1-oxyl (TEMPO), a nitroxyl radical can be a promoter in aerobic oxidation catalysis of alcohols,5,6h,15,17−19 was also investigated. However, a significant yield decrease (Fig. 4) was observed for the 1-phenylethanol oxidation, from 75.2% or 85.5% in the absence of TEMPO (for 1or 2, respectively, entry 8 or 10, Table 1) to 56.6% (1) or 58.1% (2) in its presence (entry 17 or 22, Table 1). TEMPO is also known to inhibit the further oxidation of aldehydes to carboxylic acids when this reaction occurs via a radical mechanism.19c Moreover, the previously recognised promoting effect of basic additives5,6a,b,e,17a,20 is also not observed for the present catalytic systems; in contrast, addition of 1 M aqueous solution of K2CO3 hampers the catalysis (Table 1, entries 15 and 20; Fig. 4), eventually as a result of reaction with the aqua-metal centres. The presence of HNO3 also exhibits an inhibitory effect on the acetophenone yield (Table 1, entries 16 and 21, Fig. 4), as found, e.g., for the MW-assisted oxidation of 1-phenylethanol with TBHP catalysed by Cu(II) complexes bearing Schiff base6h or 1,6-bis(2′-pyriyl)-2,5-dithiahexane ligands.6e,21 In fact, the mechanism of alcohol oxidation with TBHP does not seem to require the presence of acid as also verified6f,g in the oxidation of 1-phenylethanol with TBHP catalysed by bi- or tetra-nuclear cage-like copper(II) silsesquioxanes.
 | ||
| Fig. 4 Influence of different additives (TEMPO, K2CO3, HNO3, radical traps) on the yield of acetophenone in the MW-assisted peroxidative oxidation of 1-phenylethanol catalysed by 1 and 2. | ||
A very strong inhibition effect is observed (Fig. 4) when the peroxidative oxidation of 1-phenylethanol is carried out in the presence of either an oxygen-radical trap such as Ph2NH (Table 1, entries 18 (1) or 23 (2)) or a carbon-radical trap such as CBrCl3 (Table 1, entries 19 (1) or 24 (2)). This suggests that the oxidation reaction proceeds mainly via a radical mechanism involving both oxygen- and carbon-centred radicals.22 It may involve, e.g., the tBuO˙ radical produced in the Mn or Cu promoted decomposition of TBHP.5 It may proceed via the coordination of the alcohol substrate to an active site of the catalyst, and its deprotonation to form the alkoxide ligand, followed by a metal-centred dehydrogenation.6b,17a,20a,c,d,23
 | ||
| Fig. 5 Effect of the catalyst recycling on the yield of acetophenone from MW-assisted peroxidative oxidation of 1-phenylethanol catalysed by 1 and 2. | ||
3. Conclusions
We have achieved simple and effective syntheses of hydrosoluble mononuclear manganese(II) and tetranuclear copper(II) complexes with AHAMCs ligands, which are not soluble in low polar solvents, thus with a potential for application as heterogeneous catalysts in such media. Their structure and nuclearity are dependent on the active methylene fragment and on substituents in the aromatic part of the AHAMCs ligands, besides the metal ions. The obtained complexes act as efficient and selective heterogeneous catalyst precursors for the mild MW-assisted oxidation of secondary alcohols in solvent- and additive-free systems, thus widening the scope of heterogeneous catalytic systems suitable for MW assisted oxidative transformations of alcohols. A comparative study of their catalytic efficiency has been drawn towards different alcohol substrates.Moreover, these heterogeneous catalysts are easily recycled without considerable loss of activity. Hence, they show the same advantage, in terms of easy separation and recycling, of supported metal complex catalysts, but without requiring the use of any solid support. The approach followed in this study deserves to be further explored and extended to other oxidation catalyses and to the synthesis and catalytic applications of metal complexes of low solubility in common organic solvents.
4. Experimental
4.1. Materials and methods
All the chemicals were obtained from commercial sources and used as received. The 1H and 13C NMR spectra were recorded at room temperature on a Bruker Avance II 300 (UltraShield™ Magnet) spectrometer operating at 300.130 and 75.468 MHz for proton and carbon-13, respectively. The chemical shifts are reported in ppm using tetramethylsilane as an internal reference. The infrared spectra (4000–400 cm−1) were recorded on a BIO-RAD FTS 3000MX instrument in KBr pellets. Carbon, hydrogen and nitrogen elemental and atomic absorption (Mn and Cu) analyses were carried out by the Microanalytical Service of the Instituto Superior Técnico. All of the synthetic work was performed in air and at room temperature. The catalytic tests under microwave irradiation (MW) were performed in a focused Anton Paar Monowave 300 microwave reactor using a 10 mL capacity reaction tube with a 10 mm internal diameter, fitted with a rotational system and an IR temperature detector. Chromatographic analyses were undertaken by using a Fisons Instruments GC 8000 series gas chromatograph with a DB-624 (J&W) capillary column (DB-WAX, column length: 30 m; internal diameter: 0.32 mm), FID detector, and the Jasco-Borwin v.1.50 software. The temperature of injection was 240 °C. The initial temperature was maintained at 140 °C for 1 min, then raised 10 °C min−1 to 220 °C and held at this temperature for 1 min. Helium was used as the carrier gas. The internal standard method was used to quantify the organic products.X-ray powder diffraction patterns (XRPD) were recorded in the reflection mode using a D-8 Bruker AXS diffractometer operating at 40 kV and 40 mA, using CuKα radiation (λ = 1.5418 Å). The XRD patterns were collected at 2θ values ranging from 3° to 70° with step 0.02°. Electrospray mass spectra (ESI-MS) were run with an ion-trap instrument (Varian 500-MS LC Ion Trap Mass Spectrometer) equipped with an electrospray ion source. For electrospray ionization, the drying gas and flow rate were optimized according to the particular sample with 35 p.s.i. nebulizer pressure. Scanning was performed from m/z 50 to 1200 in methanol solution. The compounds were observed in the negative or positive mode (capillary voltage = 80–105 V).
4.2. Synthesis of complexes
:1, v/v) mixture (20 mL). The reaction mixture was heated under reflux for 3 h. A deep red precipitate was obtained. The reaction mixture was then filtered and the filtrate was rejected. The residue obtained was washed several times with acetone. The crystals of 1 suitable for X-ray structural analysis were obtained by slow evaporation of a methanol solution of the deep red solid.Yield, 27 mg, 70%. Anal. calcd for C18H34MnN8O18S2 (Mr = 769.59): C, 28.09; H, 4.45; N, 14.56. Found: C, 28.42; H, 4.61; N, 14.17%. MS (ESI) m/z: 81.5 [Mn(H2O)6]2+, 163.0 [Mn(H2O)6]+ and 267.2 (HL1a)−. IR, cm−1: 3436 ν(OH), 3103 ν(NH), 2227 ν(CN), 1645 ν(CO), 1630 δ(OH) of H2O, 1513 ν(CN).
:1 v/v) mixture (16 mL). The reaction mixture was left to slow evaporation at room temperature. After two days, greenish-black crystals of 2 were obtained.Yield, 24 mg, 74%. Anal. Calcd for C28H48Cu4N4O30S4 (Mr = 1303.10) C, 25.81; H, 3.71; N, 4.30. Found: C, 25.75; H, 3.63; N, 4.07. MS (ESI): m/z: 652.3 [Cu2(H2O)6(L2)+H+]. IR, cm−1: 3307, 3217, 3129 and 2964 ν(OH), 1666 ν(CO), 1639 ν(CO), 1628 δ(OH) of H2O, 1582 ν(CN).
4.3. X-ray measurements
The analyzed crystal was immersed in cryo-oil, mounted in a Nylon loop and measured at a temperature of 296 K. Intensity data were collected using a Bruker APEX II PHOTON 100 diffractometer with graphite monochromatic Mo-Kα (λ 0.71073) radiation. Data were collected using omega scans of 0.5° per frame and full sphere of data were obtained. Cell parameters were retrieved using Bruker SMART software and refined using Bruker SAINT24 on all the observed reflections. Absorption corrections were applied using SADABS.25 Structures were solved by direct methods by using the SHELXS-97 package and refined with SHELXL-2013.26 Calculations were performed using the WinGX System–Version 1.80.03.27 The water and the hydrazine hydrogen atoms were located from the difference Fourier map and refined isotropically with the help of distance restrains. The other hydrogen atoms were placed in calculated positions and refined by using a riding model. Least square refinements with anisotropic thermal motion parameters for all the non-hydrogen atoms were employed.4.4. General procedure for the peroxidative oxidation of alcohols
Oxidation reactions of the alcohols were carried out in sealed cylindric Pyrex tubes under focused microwave irradiation as follows: the alcohol (2.5 mmol), tert-butyl hydroperoxide (TBHP) (70% aqueous solution, 5.0 mmol) and the catalyst 1 or 2 (1–10 μmol, 0.04–0.4 mol% vs. substrate) were introduced in the tube which was then placed in the microwave reactor. In the experiments with radical traps, CBrCl3 (2.5 mmol) or NHPh2 (2.5 mmol) was added to the reaction mixture. In the experiments with other additives (TEMPO, nitric acid 1 M solution, or potassium carbonate 1 M solution), a 2.5% additive/substrate molar ratio was used. The system was stirred and irradiated (5–10 W) for 0.5–2 h at 80–150 °C. After the reaction, the mixture was allowed to cool down to room temperature. 150 μL of benzaldehyde (internal standard) and 2.5 mL of CH3CN (to extract the substrate and the organic products from the reaction mixture) were added. The obtained mixture was stirred for 10 min, filtered and then a sample (1 μL) was taken from the organic phase and analysed by GC using the internal standard method. Blank tests indicate that only traces (<0.8%) of ketone or aldehyde are generated in a metal-free system.Acknowledgements
This work has been partially supported by the Foundation for Science and Technology (FCT), Portugal [PEst-OE/QUI/UI0100/2013 and EXPL/QEQ-ERQ/2243/2013]. K.T.M., M.S. and A.V.M.N. express gratitude to FCT for the post-doc fellowships. The authors acknowledge the Portuguese NMR Network (IST-UTL Centre) for access to the NMR facility, and the IST Node of the Portuguese Network of mass-spectrometry (Dr Conceição Oliveira) for the ESI-MS measurements.References
- R. A. Sheldona and J. K. Kochi, Metal-Catalyzed Oxidations of Organic Compounds, Academic Press, New York, 1981 Search PubMed.
- Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 6th edn., 2002 Search PubMed.
- M. Hudlicky, Oxidations in Organic Chemistry, ACS monograph series, ACS, Washington, 1990 Search PubMed.
- S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234–6458 CrossRef CAS PubMed.
- Y. Y. Karabach, M. N. Kopylovich, K. T. Mahmudov and A. J. L. Pombeiro, Microwave- assisted catalytic oxidation of alcohols to carbonyl compounds, in: Advances in Organometallic Chemistry and Catalysis: The Silver/Gold Jubilee International Conference on Organometallic Chemistry Celebratory Book, ed. A. J. L. Pombeiro, Wiley, 2014, ch.18, pp. 233−245 Search PubMed.
- (a) R. A. Sheldon, Chem. Commun., 2008, 29, 3352–3365 RSC; (b) P. J. Figiel, A. Sibaouih, J. U. Ahmad, M. Nieger, M. T. Räisänen, M. Leskelä and T. Repo, Adv. Synth. Catal., 2009, 351, 2625–2632 CrossRef CAS; (c) P. Gamez, I. W. C. E. Arends, R. A. Sheldon and J. Reedijk, Adv. Synth. Catal., 2004, 346, 805–811 CrossRef CAS; (d) P. Gamez, I. W. C. E. Arends, J. Reedijk and R. A. Sheldon, Chem. Commun., 2003, 2414–2415 RSC; (e) J. S. Uber, Y. Vogels, D. van den Helder, I. Mutikainen, U. Turpeinen, W. T. Fu, O. Roubeau, P. Gamez and J. Reedijk, Eur. J. Inorg. Chem., 2007, 4197–4206 CrossRef CAS; (f) M. S. Dronova, A. N. Bilyachenko, A. I. Yalymov, Y. N. Kozlov, L. S. Shul'pina, A. A. Korlyukov, D. E. Arkhipov, M. M. Levitsky, E. S. Shubina and G. B. Shul'pin, Dalton Trans., 2014, 872–882 RSC; (g) A. N. Bilyachenko, M. S. Dronova, A. I. Yalymov, A. A. Korlyukov, L. S. Shul'pina, D. E. Arkhipov, E. S. Shubina, M. M. Levitsky, A. D. Kirilin and G. B. Shul'pin, Eur. J. Inorg. Chem., 2013, 5240–5246 CrossRef CAS; (h) A. Sabbatini, L. M. D. R. S. Martins, K. T. Mahmudov, M. N. Kopylovich, M. G. B. Drew, C. Pettinari and A. J. L. Pombeiro, Catal. Commun., 2014, 48, 69–72 CrossRef CAS PubMed; (i) R. Nasani, M. Saha, S. M. Mobin, L. M. D. R. S. Martins, A. J. L. Pombeiro, A. M. Kirillov and S. Mukhopadhyay, Dalton Trans., 2014, 9944–9954 RSC; (j) K. T. Mahmudov, M. N. Kopylovich, A. Sabbatini, M. G. B. Drew, L. M. D. R. S. Martins, C. Pettinari and A. J. L. Pombeiro, Inorg. Chem., 2014, 53, 9946–9958 CrossRef CAS PubMed; (k) N. Q. Shixaliyev, A. V. Gurbanov, A. M. Maharramov, K. T. Mahmudov, M. N. Kopylovich, L. M. D. R. S. Martins, V. G. Nenajdenko and A. J. L. Pombeiro, New J. Chem., 2014, 38, 4807–4815 RSC.
- (a) M. N. Kopylovich, Y. Y. Karabach, K. T. Mahmudov, M. Haukka, A. M. Kirillov, P. J. Figiel and A. J. L. Pombeiro, Cryst. Growth Des., 2011, 11, 4247–4252 CrossRef CAS; (b) M. N. Kopylovich, A. Mizar, M. F. C. Guedes da Silva, T. C. O. Mac Leod, K. T. Mahmudov and A. J. L. Pombeiro, Chem.–Eur. J., 2013, 19, 588–600 CrossRef CAS PubMed.
- (a) T. C. O. Mac Leod, M. N. Kopylovich, M. F. C. Guedes da Silva, K. T. Mahmudov and A. J. L. Pombeiro, Appl. Catal., A, 2012, 439–440, 15–23 CrossRef CAS PubMed; (b) M. N. Kopylovich, T. C. O. Mac Leod, M. Haukka, G. I. Amanullayeva, K. T. Mahmudov and A. J. L. Pombeiro, J. Inorg. Biochem., 2012, 115, 72–77 CrossRef CAS PubMed.
- M. N. Kopylovich, M. J. Gajewska, K. T. Mahmudov, M. F. C. Guedes da Silva, M. V. Kirillova, P. J. Figiel, J. Sanchiz and A. J. L. Pombeiro, New J. Chem., 2012, 36, 1646–1654 RSC.
- (a) A. Mizar, M. F. C. Guedes da Silva, M. N. Kopylovich, S. Mukherjee, K. T. Mahmudov and A. J. L. Pombeiro, Eur. J. Inorg. Chem., 2012, 2305–2313 CrossRef CAS; (b) M. N. Kopylovich, K. T. Mahmudov, M. Haukka, P. J. Figiel, A. Mizar, J. A. L. da Silva and A. J. L. Pombeiro, Eur. J. Inorg. Chem., 2011, 4175–4181 CrossRef CAS.
- (a) K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva, P. J. Figiel, Y. Y. Karabach and A. J. L. Pombeiro, J. Mol. Catal. A: Chem., 2010, 318, 44–50 CrossRef CAS PubMed; (b) M. N. Kopylovich, K. T. Mahmudov, M. F. C. Guedes da Silva, M. L. Kuznetsov, P. J. Figiel, Y. Y. Karabach, K. V. Luzyanin and A. J. L. Pombeiro, Inorg. Chem., 2011, 50, 918–931 CrossRef CAS PubMed.
- (a) M. N. Kopylovich, K. T. Mahmudov, A. Mizar and A. J. L. Pombeiro, Chem. Commun., 2011, 47, 7248–7250 RSC; (b) K. T. Mahmudov, A. M. Maharramov, R. A. Aliyeva, I. A. Aliyev, M. N. Kopylovich and A. J. L. Pombeiro, Anal. Lett., 2010, 43, 2923–2938 CrossRef CAS; (c) S. R. Gadjieva, T. M. Mursalov, K. T. Mahmudov, F. G. Pashaev and F. M. Chyragov, J. Anal. Chem., 2006, 61, 550–555 CrossRef; (d) K. T. Mahmudov, R. A. Aliyeva, S. R. Gadjieva and F. M. Chyragov, J. Anal. Chem., 2008, 63, 435–438 CrossRef; (e) K. T. Mahmudov, A. M. Maharramov, R. A. Aliyeva, I. A. Aliyev, R. K. Askerov, R. Batmaz, M. N. Kopylovich and A. J. L. Pombeiro, J. Photochem. Photobiol., A, 2011, 219, 159–165 CrossRef CAS PubMed.
- K. Robinson, G. V. Gibbs and P. H. Ribbe, Science, 1971, 172, 567–570 CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- (a) M. Alexandru, M. Cazacu, A. Arvinte, S. Shova, C. Turta, B. C. Simionescu, A. Dobrov, E. C. B. A. Alegria, L. M. D. R. S. Martins, A. J. L. Pombeiro and V. B. Arion, Eur. J. Inorg. Chem., 2014, 120–131 CrossRef CAS; (b) M. Sutradhar, L. M. D. R. S. Martins, M. F. C. Guedes da Silva, E. C. B. A. Alegria, C. M. Liu and A. J. L. Pombeiro, Dalton Trans., 2014, 3966–3977 RSC; (c) R. Borthakur, M. Asthana, A. Kumar, A. Koch and R. A. Lal, RSC Adv., 2013, 3, 22957–22962 RSC; (d) M. Sutradhar, L. M. D. R. S. Martins, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Appl. Catal., A, 2015, 493, 50–57 CrossRef CAS PubMed; (e) M. Sutradhar, L. M. D. R. S. Martins, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Coord. Chem. Rev., 2015 DOI:10.1016/j.ccr.2015.01.007.
- R. S. Varma, R. K. Saini and R. Dahiya, Tetrahedron Lett., 1997, 38, 7823–7824 CrossRef CAS.
- (a) R. A. Sheldon and I. W. C. E. Arends, Adv. Synth. Catal., 2004, 346, 1051–1071 CrossRef CAS; (b) W. Adam, C. R. Saha-Möller and P. A. Ganeshpure, Chem. Rev., 2001, 101, 3499–3548 CrossRef CAS PubMed; (c) M. Shibuya, Y. Osada, Y. Sasano, M. Tomizawa and Y. Iwabuchi, J. Am. Chem. Soc., 2011, 133, 6497–6500 CrossRef CAS PubMed.
- Z. Ma, L. Wei, E. C. B. A. Alegria, L. M. D. R. S. Martins, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2014, 4048–4058 RSC.
- (a) Q. Cao, L. M. Dornan, L. Rogan, N. L. Hughes and M. J. Muldoon, Chem. Commun., 2014, 50, 4524–4543 RSC; (b) J. M. Hoover, B. L. Ryland and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 2357–2367 CrossRef CAS PubMed; (c) G. Tojo and M. Fernández, Oxidation of Primary Alcohols to Carboxylic Acids: A Guide to Current Common Practice, Springer, New York, 2007 Search PubMed.
- (a) R. A. Sheldon and I. W. C. E. Arends, J. Mol. Catal. A: Chem., 2006, 251, 200–214 CrossRef CAS PubMed; (b) A. Dijksman, I. W. C. E. Arends and R. A. Sheldon, Org. Biomol. Chem., 2003, 1, 3232–3237 RSC; (c) P. J. Figiel, M. Leskelä and T. Repo, Adv. Synth. Catal., 2007, 349, 1173–1179 CrossRef CAS; (d) J. U. Ahmad, P. J. Figiel, M. T. Räisänen, M. Leskelä and T. Repo, Appl. Catal., A, 2009, 371, 17–21 CrossRef CAS PubMed; (e) G. Yang, W. Zhu, P. Zhang, H. Xue, W. Wang, J. Tian and M. Songa, Adv. Synth. Catal., 2008, 350, 542–546 CrossRef CAS; (f) L. Lin, M. Juanjuan, J. Liuyan and W. Yunyang, J. Mol. Catal. A: Chem., 2008, 291, 1–4 CrossRef PubMed; (g) L. Lin, M. Juanjuan, J. Liuyan and W. Yunyang, Catal. Commun., 2008, 9, 1379–1382 CrossRef CAS PubMed; (h) S. Striegler, Tetrahedron, 2006, 62, 9109–9114 CrossRef CAS PubMed.
- R. R. Fernandes, J. Lasri, M. F. C. Guedes da Silva, J. A. L. da Silva, J. J. R. Frausto da Silva and A. J. L. Pombeiro, Appl. Catal., A, 2011, 402, 110–120 CrossRef CAS PubMed.
- (a) L. M. Slaughter, J. P. Collman, T. A. Eberspacher and J. I. Brauman, Inorg. Chem., 2004, 43, 5198–5204 CrossRef CAS PubMed; (b) J. A. Howard, in: Free Radicals, ed. J. K. Kochi, Wiley, New York, vol. II, 1973, p. 3 Search PubMed; (c) R. E. Huie and C. L. Clifton, Int. J. Chem. Kinet., 1989, 21, 611–619 CrossRef CAS; (d) I. N. Moiseeva, A. E. Gekham, V. V. Minin, G. M. Larin, M. E. Bashtanov, A. A. Krasnovskii and I. I. Moiseev, Kinet. Catal., 2000, 41, 170–177 CrossRef; (e) J. M. Mattalia, B. Vacher, A. Samat and M. Chanon, J. Am. Chem. Soc., 1992, 114, 4111–4119 CrossRef CAS; (f) T. F. S. Silva, M. F. C. Guedes da Silva, G. S. Mishra, L. M. D. R. S. Martins and A. J. L. Pombeiro, J. Organomet. Chem., 2011, 696, 1310–1318 CrossRef CAS PubMed; (g) T. F. S. Silva, T. C. O. Mac Leod, L. M. D. R. S. Martins, M. F. C. Guedes da Silva and A. J. L. Pombeiro, J. Mol. Catal. A: Chem., 2013, 367, 52–60 CrossRef CAS PubMed; (h) T. F. S. Silva, L. M. D. R. S. Martins, M. F. C. Guedes da Silva, A. R. Fernandes, A. Silva, P. M. Borralho, S. Santos, C. M. P. Rodrigues and A. J. L. Pombeiro, Dalton Trans., 2012, 41, 12888–12897 RSC; (i) A. I. F. Venâncio, M. F. C. Guedes da Silva, L. M. D. R. S. Martins and A. J. L. Pombeiro, Organometallics, 2005, 24, 4654–4665 CrossRef; (j) A. I. F. Venâncio, M. L. Kuznetsov, M. F. C. Guedes da Silva, L. M. D. R. S. Martins, J. J. R. Fraústo da Silva and A. J. L. Pombeiro, Inorg. Chem., 2002, 41, 6456–6467 CrossRef PubMed.
- (a) M. V. N. de Souza, Mini-Rev. Org. Chem., 2006, 3, 155–165 CrossRef CAS; (b) T. Vogler and A. Studer, Synthesis, 2008, 1979–1993 CAS; (c) A. Dijksman, I. W. C. E. Arends and R. A. Sheldon, Org. Biomol. Chem., 2003, 1, 3232–3237 RSC; (d) B. L. Ryland and S. S. Stahl, Angew. Chem., Int. Ed., 2014, 53, 8824–8838 CrossRef CAS PubMed.
- Bruker, APEX2 & SAINT, Bruker, AXS Inc., Madison, Wisconsin, USA, 2004 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical data for compounds NaHL1 and Na2H2L2. Crystallographic and selected structural details are listed in Tables S1 and S2. CCDC 1023179 and 1023180. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra02667a |
| This journal is © The Royal Society of Chemistry 2015 |