
Multi-bond forming and iodo-selective base-promoted homolytic aromatic substitution†
Kye-Simeon Masters*
Chemistry, Physics and Mechanical Engineering School (CPME), Queensland University of Technology (QUT), Brisbane, Queensland, Australia. E-mail: kye.masters@qut.edu.au
First published on 12th March 2015
Abstract
Base-promoted homolytic aromatic substitution (BHAS) has been applied as a means to effect multi-bond forming reactions. Using the dioxepine framework to illustrate the concept, BHAS was used to rapidly access regio-defined polycycles in merely 2 or 3 steps from iodophenol. Both liquid and solid arenes of an electronically-diverse nature were used as the coupling partner/solvent. DMSO additive was found to promote this multi-BHAS reaction. A pathway consistent with these observations is suggested, this hypothesis was exploited to further develop an iodo-selective BHAS reaction.
Introduction
Since Ullmann's copper-mediated synthesis of the biaryl bond, this most crucial1 of carbon–carbon connections has been the subject of extensive synthetic advances. As a result, most methods which have been developed for the construction of multiple biaryl bonds in a single operation have hitherto relied on transition metal-catalysis. Arguably the most promising metal-free alternative2 to established transition metal-mediated methods of cross-catalysis3 and C–H functionalization4 is the recently-discovered Base-promoted Homolytic Aromatic Substitution (BHAS). BHAS features the use of a tert-butoxide base5 alongside a sub-stoichiometric amount of diamine (or other) initiator to generate aryl radical species6a and is a hot topic in the contemporary iteration of a ‘radical renaissance’.6b
Multi-bond forming reactions7 provide expedient, selective and often high-yielding access to desirable targets and synthetic intermediates with minimal expenditure of chemical resources. Early BHAS work has demonstrated its potential to generate multiple C–C bond formations via twofold substitutions5b,c,8,9 and showcased interesting cyclizations.10 Initial multi-BHAS examples have been restricted to substitution of two halide functional handles upon a single arene core with benzene as coupling partner (Scheme 1); further advances appear not to have been made on this aspect of BHAS methodology to date. In this work, a multi-bond forming methodology has been developed to effect both an intramolecular BHAS to a tethered arene and an intermolecular BHAS with the arene solvent/reactant. Uniquely for multi-bond forming BHAS reactions: (i) the two reactive halo-functionalities of the multi-bond forming reaction are located on separate arenes; (ii) the H-arene is not limited to benzene; and (iii) conditions have been found for effective control of iodo- (over bromo-) arene selectivity. Mechanistic reasoning suggests that this reaction may comprise elements of each ‘domino’ and ‘multi-bond forming transformation of independent’ reactions.7d
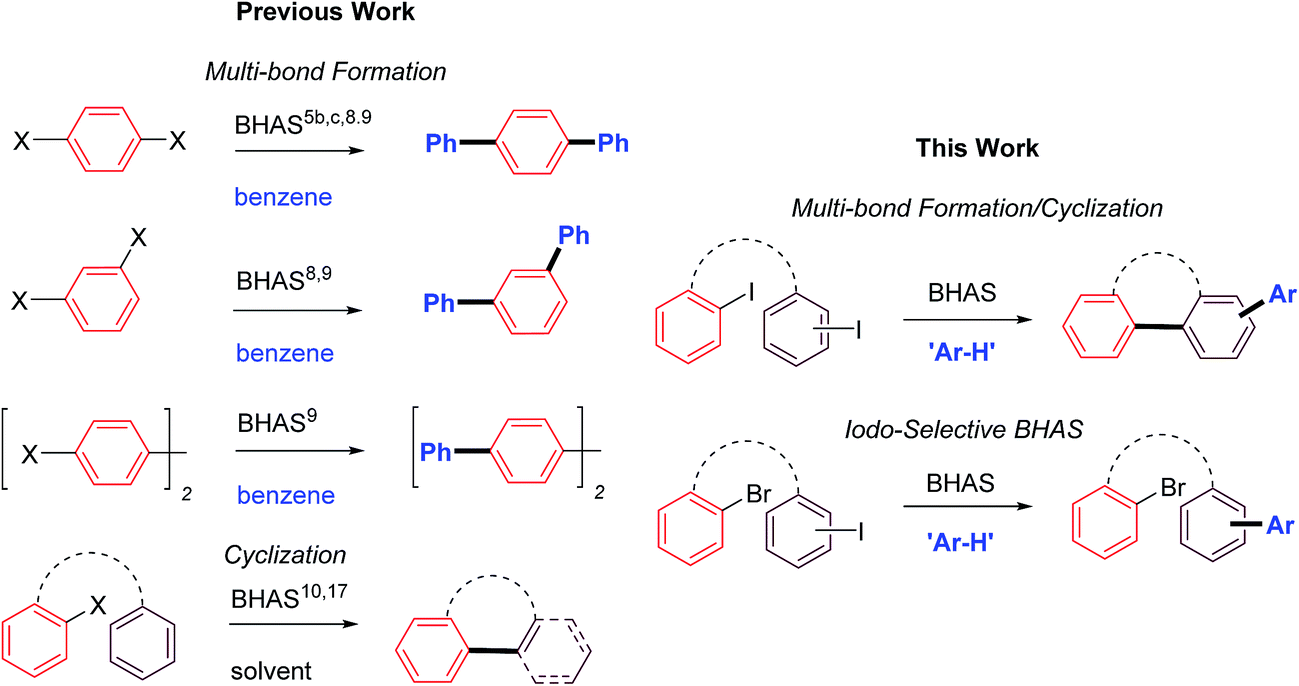 | ||
| Scheme 1 Existing and new classes of BHAS transformation. | ||
Break-through publications on transition metal-free, alkali-metal tert-butoxide-promoted C–H substitutions have been contributed by Itami and co-workers5a and the groups of Hayashi and Shirakawa,5b Lei and Kwong,5c and Shi.5d The literature has since seen a rapidly-increasing interest in BHAS, which has demonstrated that it may be promoted photolytically9 or with a variety of additives11 to effect cyclization,10 arene–arene coupling,12 and Heck-like substitution.13 Studer and Curran described6a the mechanism of this homolytic aromatic substitution as a radical chain-reaction, where the electron is passed from reactant to reactant14 – i.e. the ‘electron is a catalyst’.6c The source of the initial electron (the initiation step) in this reaction is the subject of some debate – several suggestions have been made.15
Inspired by both the ‘lactone method’16 and the innovative early BHAS cyclization work of Charette and co-workers,10a the ‘acetal method’ is a strategy for the synthesis of 2,2′(ortho,ortho-)biphenols via dibenzo[1,3]dioxepines utilizing BHAS for the key ring-closing reaction.17 Expanding on this initial work, non-symmetrical aryl-substituted dibenzo[1,3]dioxepines were chosen as targets to explore BHAS in the context of multi-bond forming reactions.
Results
Synthesis of non-symmetrical and symmetrical di(haloaryloxy)methylene ethers by the previously-reported method17b saw conversion of 4-iodophenol to the mixed O,S-methylene diether, 2 (Scheme 2), which was substituted in a two-stage one-pot reaction with sulfuryl chloride and then 2-bromophenol, delivering bromoaryl-iodoarene 4 in 84% yield (over 2 steps). This sequence with 2-iodophenol led to 2,4′-diiodoaryloxy ether 6 (79% over 2 steps). 2-Iodophenol was also converted to 2,3′-diiodoaryloxy methane 10 (by way of 8, 87%, two steps) and to bis-(2-iodoaryloxy)methane 7 (93%, one step). | ||
| Scheme 2 Dihaloarene substrates for multi-bond forming BHAS. Conditions: (a) NaH, then ClCH2SCH3, DMF, r.t.; (b/i) SO2Cl2, CH2Cl2, r.t., (ii) halophenol, NaH, DMF, r.t., then mixture from (b/i) in DMF; (c) NaH, CH2I2, DMF, r.t. | ||
The cascade reaction was explored utilizing 2-bromophenoxy-4′-iodophenoxymethane, 4, with the conditions optimised by Charette18 and previously successful for the simple cyclization to dioxepines: potassium tert-butoxide (6.0 equiv., i.e. 3.0 equivalents per halogen moiety), 1,10-phenanthroline (80%) at 140 °C with benzene and pyridine as substrate and solvent, respectively (ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7, entry 1, Table 1). These initial conditions proved to be unsatisfactory, as only traces of desired product 11 were observed. Using benzene as both solvent/reactant delivered the multi-BHAS compound in 23% yield, albeit as part of a complex mixture of at least four identifiable products (11–14). These (by)products result from coupling and 7-ortho-cyclization (11),17b and de-halogenation (12),19 then 6-ipso-cyclization10a then C → O transposition20 and elimination of formaldehyde (to 13),17b and aryl alkoxylation by nucleophilic or radical aromatic substitution (14).19 Change of the substrate to diiodide 6 gave an improved ratio between the products (entry 3), but the yield of 11 remained low (21%).
:7, entry 1, Table 1). These initial conditions proved to be unsatisfactory, as only traces of desired product 11 were observed. Using benzene as both solvent/reactant delivered the multi-BHAS compound in 23% yield, albeit as part of a complex mixture of at least four identifiable products (11–14). These (by)products result from coupling and 7-ortho-cyclization (11),17b and de-halogenation (12),19 then 6-ipso-cyclization10a then C → O transposition20 and elimination of formaldehyde (to 13),17b and aryl alkoxylation by nucleophilic or radical aromatic substitution (14).19 Change of the substrate to diiodide 6 gave an improved ratio between the products (entry 3), but the yield of 11 remained low (21%).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry, substrate | Solvent | Conc. (M) | DMSO (equiv.) | Time (h) | 11 (%) | 12 (%) | 13 (%) | 14 (%) |
| a Conditions: 1,10-phen. (80 mol%), KOtBu (6.0 equiv.), Ar–H (20–40 equiv.), DMSO (5.0 equiv.), microwave irradiation (MWI), 140 °C. All yields are isolated. *1,10-Phenanthroline was excluded. ND = none detected. | ||||||||
| 1, 4 | Pyr./PhH (7:1) |
0.25 | — | 120 | ND | 24 | ND | ND |
| 2, 4 | C6H6 | 0.25 | — | 240 | 23 | 5 | 12 | 50 |
| 3, 6 | C6H6 | 0.125 | — | 120 | 21 | 12 | 11 | 30 |
| 4, 6 | C6H6 | 0.125 | 5.0 | 240 | 59 | 19 | 14 | ND |
| 5, 6* | C6H6 | 0.125 | 5.0 | 240 | ND | ND | ND | ND |

Rossi and co-workers have discovered that DMSO improves BHAS yields under photoactivation at room temperature.9 Happily, when a small amount of DMSO was included as an additive with the conditions above (5.0 equiv., entry 4), the outcome of this thermally-promoted reaction was also greatly improved in terms of yield and selectivity. DMSO additive increased the isolated yield of cascade product 11 from 59%, and prevented generation of alkoxy-substituted byproduct 14. A control experiment with DMSO in the absence of 1,10-phenanthroline showed that DMSO alone is not capable of promoting the reaction (entry 5).
With satisfactory conditions determined, the cascade BHAS was then applied to cyclization/coupling with several electronically-diverse arene partners. Complementing the usual use of liquid arenes (benzene and 1,4-difluorobenzene) some of these were solids at room temperature (1,4-dimethoxybenzene, pyrazine, naphthalene, Scheme 3). Physical separation of the base and initiator in distinct layers of the reaction vessel prior to heating ensured that the reaction did not commence until the arene was liquefied (see Fig. 1, ESI†). Diiodides 6 and 7 produced 2- and 4-aryl-substituted dioxepines 15–18 and 19–24a/b in moderate to good yields (50–71%, Scheme 3), and meta-iodo substrate 10 gave a mixture of each of the two possible products (24a/b). Coupling of symmetrical diiodide 7 with benzene to afford 20 was notably poor in yield (38%-unsubstituted dioxepine was consistently isolated in 12–14% yield). These yields are satisfactory when considered in light of the two discrete C–C bond forming events that must take place. All 4 possible Ph-substituted isomers (11, 20, 24a and 24b) were isolated and characterized.
 | ||
| Scheme 3 Multi-bond forming to aryl-substituted dibenzo[1,3]dioxepines. Conditions: 1,10-phen. (80 mol%), KOtBu (6.0 equiv.), Ar–H (10–30 equiv.), DMSO (5.0 equiv.), MWI, 140 °C, 4 h. | ||
Although multiple equivalents of arene are used in this method, the low cost of these coupling partners makes their use nonetheless advantageous. In contrast with typical cross-coupling substrates to form biaryls (aryl-zincs, -boronic acids, etc.), standard H-arenes not requiring preactivation or in situ preparation, are cheap,21 non-toxic, do not require careful preparation, and are readily to hand in most chemistry laboratories.
Discussion
A reaction pathway accounting for observations and isolated byproducts is proposed (Scheme 4). EPR studies have demonstrated the generation of radicals in BHAS reactions,22a,b and a recent report by Jutand and Lei has investigated the reaction with cyclic voltametry.22c Single-electron-transfer (SET) is thought to initially result in a radical anion located upon the haloarene – the observed non-reactivity of substrate 6 in the presence of 1,4-dinitrobenzene an established radical anion scavenger,23 further supports the role of a radical anion (see Scheme 3). | ||
| Scheme 4 Proposed mechanism for the multi-BHAS to form 11–14. | ||
The multi-BHAS from diiodoaryl 6 exhibits a uniform arene-coupling outcome at the (formerly iodo-bearing) C-(para) position, whereas there are at least four modes of reaction for the C-(ortho) position, as evidenced by the products isolated (their distribution varies with reaction conditions, see Table 1). These results suggest that in addition to the most straightforward ortho-σ-radical pathway to 11 via initially-cyclised 26,24 there is some degree of independently-operating pathway where initial substitution is at the apparently more reactive para-iodo-substituted position of 6 to form radical intermediate 25 then 28. This cyclohexadienyl radical undergoes rapid transfer of its (extremely acidic) proton25,22a then either intramolecular ET (to 29 then 11) or intermolecular ET to another iodoarenyl moiety on a different molecule (6 or 30), and continuation of the chain process. Intermediate ortho-σ-type radical 29 can apparently follow one of four divergent pathways: pathway 1 involves 7-ortho cyclization to also result in 11, pathway 2 sees 6-ipso cyclization, then C → O transposition and loss of ‘CH2O’ to (relatively) stable phenoxyl radical then phenol 13, in pathway 3 the ortho-σ-radical abstracts hydrogen or is quenched to anion with another free electron in situ then protonated, giving 12. Lastly, butoxyl substituted 14 may be generated by either a radical19a or nucleophilic aromatic substitution11m,19b pathway, from 29 or 30, respectively (or perhaps both).19c Having previously established that 2-bromo substrate 4 gave more of t-butyl ether 14 than 2-iodo 6 (see Table 1), the latter pathway is suggested. It is likely that the pathways from substrates 7 and 10 are similarly complex.
The role of DMSO co-solvent in promoting formation of 11 and suppressing the side-reactions to, e.g. tert-butoxyl substituted arene 14, is not yet clear, but may be to enhance the domino pathway over the ‘multiple independent reaction’ pathway. Visually, these reactions exhibited a more rapid formation of the dark coloration linked to initiation, and enhanced radical-initiation is a strong possibility.
The addition of KOt-Bu to DMSO rapidly forms an equilibrium mixture with t-BuOH/dimsyl potassium superbase (KCH2SOCH3, 34, Scheme 5).26 In the presence of 1,10-phenanthroline, the potassium dimsyl salt may be chelating to form a potassium amide superbase 38 analogous to 1,3-diaminopropane which makes KAPA (potassium 3-aminopropy-1-amide, 36)27a still more basic than potassium dimsyl by a factor of 105 to 106.27b Such a species may undergo an inner-sphere electron-transfer to form phen˙− and tBuO˙ as suggested by Lei and Jutand,22c or perhaps promote the formation of the attractive super electron donor 39, proposed by Tuttle and Murphy, which requires deprotonations of 1,10-phenanthroline, 37.15a To present a further possibility, Wilden and co-workers have recently described a dynamic equilibrium between ‘nearly-covalent’ KOt-Bu (29), its charge-separated form (39a), and a charge separated form with a loosely-bound electron (40a), which alone can provide for the initial SET to the haloarene at elevated temperatures (160 °C).28 It may be the case that a shifting in the equilibrium position to favor the latter of these species (39 and 40) is enhanced in the presence of cation-stabilising DMSO. An alternative is that the dimsyl potassium superbase, which is a strong electrolyte itself,29 also has a loosely-bound electron form which can be donated. Regardless, the effect of DMSO addition is enhancement of the pathway to 11, outcompeting that to 14.
 | ||
| Scheme 5 Possible roles of DMSO in promoting the BHAS initiation. | ||
Given the postulated mechanism above, it was hoped that the reaction conditions could be tuned so as to block reactivity of the 2-halo position upon the initially-used bromoaryl species 4. Selectivity for the p-iodo functionality BHAS was convincingly obtained by excluding the DMSO additive, decreasing the reaction temperature to 100 °C, and shortening the duration of the reaction to 60 minutes, conditions which maintained the 2′-bromoaryl functionality intact (43–46, 50–86% yield). Chemo-/regio-selective control is so far poorly controllable in the BHAS reaction, but could allow for sequential BHAS/BHAS and BHAS/TM-catalysis coupling sequences, e.g. in divergent synthesis. The scope of para-iodo-selective coupling partners matched that of the multi-BHAS (Scheme 6).
 | ||
| Scheme 6 Iodoarene-selective BHAS in the presence of a bromoarene moiety. Conditions: 1,10-phen. (40 mol%), KOtBu (3.0 equiv.), Ar–H (20–40 equiv.), MWI, 100 °C, 1 h. | ||
Conclusion
The BHAS reaction should be considered a synthetic option when multi-bond forming reactions are desired for rapid, modular generation of polycycles or other compounds with multiple biaryl connections. A multi-BHAS approach can work well, eschewing the use of transition-metals. Iodo-selective conditions were also established to allow attachment of only one arene to the scaffold, leaving the bromoarene intact for a host of potential subsequent reaction methods. The multi-bond and stepwise BHAS methods make use of relatively cheap and environmentally benign substrates/reagents, and thus holds promise for efficient transformations in chemical Industry, decreasing costs ‘on scale’ and precluding trace amounts of toxic transition metals. Continuation of studies focusing on the further scope of the multi-bond forming BHAS and role of DMSO is currently underway in the laboratory.Experimental section
General experimental
Chemicals and solvents were used as commercially supplied without further purification, unless otherwise noted. Potassium tert-butoxide (95%), 1,10-phenanthroline (99%), sodium hydride (60% dispersion in mineral oil), dimethyl sulfoxide (Reagentplus ≥99.5% purity), chloromethyl methylsulfide (95%), 1,4-dimethoxybenzene (99%), pyrazine (≥99%), naphthalene (99+%), sulfuryl chloride (97%) and dimethylformamide (99.5%, anhydrous) were supplied by Aldrich. Benzene (analytical reagent grade, obtained from Chemsupply Australia) was dried over anhydrous molecular sieves prior to use. Optimal results for this methodology required the storage of potassium tert-butoxide in a desiccator, and commercially-available 1,10-phenanthroline was dried of water prior to use by addition of dry toluene azeotroping out on the rotavap (repeat ×2). Microwave reactions were performed in a Biotage Initiator+ with a maximum power of 400 W, reactions were in new vials with securely-sealed (crimped) microwave tubes. Column chromatography was performed on Davisil (LC60A, 40–63 μm Grace). Preparative HPLC was performed on an Agilent HPLC with a C18 column (25 × 200 mm) using tetrahydrofuran and water (both HPLC grade, Merck). NMR data were recorded on a Varian Infinity-Plus 400 spectrometer (1H at 400 MHz; 13C at 100 MHz), and resonances are reported in terms of chemical shift (δ) in parts per million (ppm) referenced to the solvent peak; coupling constants (J) are given in Hertz (Hz) and the number of protons per signal as nH. Splitting is reported as br. = broad, s = singlet, d = doublet and m = multiplet.General procedures (1–4)
Preparation of specific compounds
:1, 150 mL, ×2). The organics were washed with H2O (200 mL × 2), brine (50 mL) and dried over sodium sulfate. Filtration and removal of the volatiles by rotary evaporation under diaphragm pump vacuum (to 30 Mbar) followed, then the crude was purified by column chromatography on silica gel with hexane/CH2Cl2 (9:1). Isolated as a colourless semi-solid, 13.14 g, 46.9 mmol, 94% yield. MP: ca. room temperature; 1H NMR (400 MHz, CDCl3): δ = 7.58 (dt, J = 9.0, 2.0 Hz, 2H), 6.73 (dd, J = 9.0, 2.3 Hz, 2H), 5.11 (s, 2H), 2.23 (s, 3H); 13C NMR (101 MHz, CDCl3): δ = 156.8, 138.3, 118.3, 84.2, 72.4, 14.6; LRMS (+EI, GCMS): found 280.0; HRMS (+EI): calculated [C8H9IOS]˙+: 279.9419; found:279.9419.Stage 1. Prepared in a 2-stage pseudo-1-pot reaction from compound 2 and 2-bromophenol by a modification of the method of Masters and Bräse (two stages, 89%).17 To a stirred solution of [(4-iodophenoxy)methyl] (methyl)sulfane 2 (1.40 g, 5.00 mmol) in dry CH2Cl2 (25 mL) under argon in a 100 mL round-bottomed flask held at −5 °C in a salt/ice bath was added dropwise sulfuryl chloride (410 mL, 5.125 mmol, 1.025 equiv.). The ice bath was removed and the reaction mixture was stirred up to room temperature over 60 minutes, becoming pale-yellow. After this time the volatiles were removed by rotary evaporation under diaphragm pump vacuum (to 30 Mbar; this should be done with a rotary evaporator in a fume-cupboard). The crude product is invariably obtained with complete conversion and in quantitative yield, and can be checked if desired (1H-NMR, CDCl3). This crude material was taken up in dry, non-hydrolyzed or amine-free DMF and added directly to the deprotonated phenols in stage 2.
Stage 2. To a stirred solution of 2-bromophenol (951 mg, 5.5 mmol, 1.1 equiv.) in dry, non-hydrolyzed or amine-free DMF (27.5 mL) under argon and at 0 °C was added sodium hydride (240 mg, 6.0 mmol, 1.2 equiv., 60% dispersion in mineral oil) and the mixture stirred for 30 minutes with warming to room temperature. After re-cooling to 0 °C, the aryloxymethylenechloride from stage 1 was added dropwise as a solution in DMF (5.0 mL, 2 mL washings). After stirring overnight, the reaction mixture was heated at 50 °C for 4 hours, then poured into H2O (70 mL) and extracted with CH2Cl2/hexanes (1
:1, 50 mL, ×2). The combined organics were washed with H2O (50 mL × 2), then brine (20 mL) and dried over sodium sulfate. Filtration and removal of the volatiles by rotary evaporation under diaphragm pump vacuum (to 30 Mbar) followed, then the crude was purified by column chromatography on silica gel with hexane/CH2Cl2 (9:1). Isolated as a colourless oil which solidified overnight, recrystallization from MeOH/CH2Cl2 yielded the compound as colourless needles, 1.80 g, 4.45 mmol, 89% yield. MP: 40–42 °C; 1H NMR (400 MHz, CDCl3): δ = 7.63–7.57 (m, 2H), 7.54 (dd, J = 1.6, 7.8 Hz, 1H), 7.30–7.26 (m, 1H), 7.23–7.16 (m, 1H), 6.98–6.85 (m, 3H), 5.74 (s, 2H); 13C NMR (101 MHz, CDCl3): δ = 156.8, 153.2, 138.4, 133.6, 128.5, 124.0, 118.8, 116.7, 113.1, 91.4, 85.3; LRMS (+EI, GCMS): found 403.9; HRMS (+EI): calculated [C13H10BrIO2]˙+: 403.8903; found: 403.8909.
:1). Isolated as a colourless solid, recrystallization from MeOH/CH2Cl2 yielded the compound as colourless needles, 5.18 g, 93% yield. MP: 94–95 °C; 1H NMR (400 MHz, CDCl3) δ = 7.79 (d, J = 7.8 Hz, 2H), 7.39–7.32 (m, 2H), 7.28 (s, 2H), 6.81 (t, J = 7.6 Hz, 2H), 5.82 (s, 2H); 13C NMR (101 MHz, CDCl3): δ = 155.7, 139.6, 129.7, 124.4, 115.5, 91.7, 87.1; LRMS (+EI, GCMS): found 451.9; HRMS (+EI): calculated [C13H10I2O2]˙+: 451.8765; found: 451.8768.:3, respectively). Further separated with CH2Cl2 in hexane (gradient, 12–20%) to deliver 23a. The earlier-eluting fraction was subjected to chromatography with toluene in hexane fraction (gradient, 15–30%) to deliver the 2-naphthyl isomer 23b, which was not able to be completely separated from a coupled but not cyclized compound with a mass of 326 (as determined by GCMS).:4.Acknowledgements
This work was supported by a Vice-Chancellor's Research Fellowship from the Queensland University of Technology. Mass spectra were acquired by Ms Anithahini (Anitha) Jeyasingham with support from Prof. Stephen Blanksby and QUT's Central Analytical Facility (CARF). K.-S. M. acknowledges valuable mentoring from Prof. Steven Bottle and Prof. Stefan Bräse.References
- J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1469 CrossRef CAS PubMed.
- (a) X. Zheng, L. Yang, W. Du, A. Ding and H. Guo, Chem.–Asian J., 2014, 9, 439–442 CrossRef CAS PubMed; (b) T. Kawamoto, A. Sato and I. Ryu, Org. Lett., 2014, 16, 2111–2113 CrossRef CAS PubMed.
- A. de Meijere and F. Diederich, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed.
- (a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS PubMed; (b) B. J. Li, S. D. Yang and Z. J. Shi, Synlett, 2008, 5, 949 Search PubMed; (c) O. Daugulis, H. Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS PubMed; (d) G. P. McGlacken and L. M. Bateman, Chem. Soc. Rev., 2009, 38, 2447–2464 RSC; (e) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed; (f) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed; (g) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236–10254 CrossRef CAS PubMed.
- (a) S. Yanagisawa, K. Ueda, T. Taniguchi and K. Itami, Org. Lett., 2008, 10, 4673–4676 CrossRef CAS PubMed; (b) E. Shirakawa, K.-I. Itoh, T. Higashino and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 15537–15539 CrossRef CAS PubMed; (c) W. Liu, H. Cao, H. Zhang, H. Zhang, K. H. Chung, C. He, H. Wang, F. Y. Kwong and A. Lei, J. Am. Chem. Soc., 2010, 132, 16737–16740 CrossRef CAS PubMed; (d) C.-L. Sun, H. Li, D.-G. Yu, M. Yu, X. Zhou, X.-Y. Lu, K. Huang, S.-F. Zheng, B.-J. Li and Z.-J. Shi, Nat. Chem., 2010, 2(12), 1044–1049 CrossRef CAS PubMed; (e) S. Yanagisawa and K. Itami, ChemCatChem, 2011, 3, 827–829 CrossRef CAS.
- (a) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2011, 50, 5018–5022 CrossRef CAS PubMed; (b) C. R. J. Stephenson, A. Studer and D. P. Curran, Beilstein J. Org. Chem., 2013, 9, 2778–2780 CrossRef PubMed; (c) A. Studer and D. P. Curran, Nat. Chem., 2014, 6(9), 765–773 CrossRef CAS PubMed.
- (a) L. F. Tietze and U. Beifuss, Angew. Chem., Int. Ed., 1993, 32, 131–163 CrossRef; (b) L. F. Tietze, G. Brasche and K. M. Gericke, Domino Reactions in Organic Synthesis, Wiley-VCH, Weinheim, 1st edn, 2006 Search PubMed; (c) L. Albrecht, H. Jiang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 8492–8509 CrossRef CAS PubMed; (d) N. J. Green and M. S. Sherburn, Aust. J. Chem., 2013, 66, 267–283 CrossRef CAS.
- (a) 1,4-Dihalobenzenes: S. Sharma, M. Kumar, V. Kumar and N. Kumar, Tetrahedron Lett., 2013, 54, 4868–4871 CrossRef CAS; (b) Y. Cheng, X. Gu and P. Li, Org. Lett., 2013, 15, 2664–2667 CrossRef CAS PubMed . See also ref. 5b, c and 9.
- M. E. Budén, J. F. Guastavino and R. A. Rossi, Org. Lett., 2013, 15, 1174–1177 CrossRef PubMed.
- (a) D. S. Roman, Y. Takahashi and A. B. Charette, Org. Lett., 2011, 13, 3242–3245 CrossRef CAS PubMed; (b) C.-L. Sun, Y.-F. Gu, W.-P. Huang and Z.-J. Shi, Chem. Commun., 2011, 47, 9813–9815 RSC; (c) M. Rueping, M. Leiendecker, A. Das, T. Poisson and L. Bui, Chem. Commun., 2011, 47, 10629–10631 RSC; (d) B. S. Bhakuni, A. Kumar, S. J. Balkrishna, J. A. Sheikh, S. Konar and S. Kumar, Org. Lett., 2012, 14, 2838–2841 CrossRef CAS PubMed; (e) S. De, S. Ghosh, S. Bhunia, J. A. Sheikh and A. Bisai, Org. Lett., 2012, 14, 4466–4469 CrossRef CAS PubMed; (f) C.-L. Sun, Y.-F. Gu, B. Wang and Z.-J. Shi, Chem.–Eur. J., 2011, 17, 10844–10847 CrossRef CAS PubMed; (g) Y. Wu, S. M. Wong, F. Mao, T. L. Chan and F. Y. Kwong, Org. Lett., 2012, 14, 5306–5309 CrossRef CAS PubMed.
- (a) Bathophenanthroline; ref. 4b; (b) DMEDA: ref. 4c; (c) Phenanthroline: ref. 4d; (d) Macrocyclic aromatic pyridone pentamer: H. Zhao, J. Shen, J. Guo, R. Ye and H. Zeng, Chem. Commun., 2013, 49, 2323–2325 RSC; (e) Stable zwitterionic radical: G. P. Yong, W. L. She, Y. M. Zhang and Y. Z. Li, Chem. Commun., 2011, 47, 11766–11768 RSC; (f) Free-base porphyrin: Y. S. Ng, C. S. Chan and K. S. Chan, Tetrahedron Lett., 2012, 53, 3911–3914 CrossRef CAS; (g) Proline: K. Tanimoro, M. Ueno, K. Takeda, M. Kirihata and S. Tanimori, J. Org. Chem., 2012, 77, 7844–7849 CrossRef CAS PubMed; (h) Ethylene glycol: Y. Wu, S. M. Wong, F. Mao, T. L. Chan and F. Y. Kwong, Org. Lett., 2012, 14, 5306–5309 CrossRef CAS PubMed; (i) Simple alcohols: W. Liu, F. Tian, X. Wang, H. Yu and Y. Bi, Chem. Commun., 2013, 49, 2983–2985 RSC; (j) p-Toluenesulfonohydrazide: Q. Song, D. Zhang, Q. Zhu and Y. Xu, Org. Lett., 2014, 16, 5272–5274 CrossRef CAS PubMed; (k) Bis(imino)pyridines: S. A. X. Liu, H. Li, C. He and Y. Mu, Asian J. Org. Chem., 2013, 2, 857–861 CrossRef; (l) Mixed alkoxides: W. Lui, L. Xu and Y. Bi, RSC Adv., 2014, 4, 44943–44947 RSC; (m) Y. Wu, P. Y. Choy and F. Y. Kwong, Asian J. Org. Chem., 2014, 3, 1262–1265 CrossRef CAS.
- (a) Y. Qiu, Y. Liu, K. Yang, W. Hong, Z. Li, Z. Wang, Z. Yao and S. Jiang, Org. Lett., 2011, 13, 3556–3559 CrossRef CAS PubMed; (b) O. Vakuliuk, B. Koszarna and D. T. Gryko, Adv. Synth. Catal., 2011, 353, 925–930 CrossRef CAS; (c) S. Castro, J. J. Fernandez, R. Vicente, F. J. Fananas and F. Rodriguez, Chem. Commun., 2012, 48, 9089–9091 RSC; (d) Y. S. Ng, C. S. Chan and K. S. Chan, Tetrahedron Lett., 2012, 53, 3911–3915 CrossRef CAS; (e) H. Liu, B. Yin, Z. Gao, Y. Li and H. Jiang, Chem. Commun., 2012, 48, 2033–2035 RSC; (f) Y. Cheng, X. Gu and P. Li, Org. Lett., 2013, 15, 2664–2667 CrossRef CAS PubMed; (g) Y. Yoshimi, H. Kanai, K. Nishikawa, Y. Ohta, Y. Okita, K. Maeda and T. Morita, Tetrahedron Lett., 2013, 54, 2419–2422 CrossRef CAS.
- (a) J. F. Guastavino, M. E. Budén and R. A. Rossi, J. Org. Chem., 2014, 79, 9104–9111 CrossRef CAS PubMed; (b) E. Shirakawa, X. Zhang and T. Hayashi, Angew. Chem., Int. Ed., 2011, 50, 4671–4674 CrossRef CAS PubMed ; see also ref. 10c and f.
- (a) A. Studer and M. Bossart, in Radicals in Organic Synthesis, ed. P. Renaud and M. P. Sibi, Wiley-VCH Verlag, Weinheim, 1st edn, 2001, vol. 2, p. 62 Search PubMed; (b) R. Bolton and G. H. Williams, Chem. Soc. Rev., 1986, 15, 261–289 RSC; (c) J. Fossey, D. Lefort and J. Sorba, Free Radicals in Organic Chemistry, Wiley, Chichester, 1995, ch. 14, pp. 166–180 Search PubMed; (d) A. L. J. Beckwith, V. W. Bowry, W. R. Bowman, E. Mann, J. Parr and J. M. D. Storey, Angew. Chem., Int. Ed., 2004, 116, 97–100 (Angew. Chem., Int. Ed., 2004, 43, 95–98) CrossRef; (e) A. N. Hancock and C. H. Schiesser, Chem. Commun., 2013, 49(85), 9892–9895 RSC.
- (a) S. Zhou, G. M. Anderson, B. Mondal, E. Doni, V. Ironmonger, M. Kranz, T. Tuttle and J. A. Murphy, Chem. Sci., 2014, 5, 476–482 RSC; (b) A. Dewanji, S. Murarka, D. P. Curran and A. Studer, Org. Lett., 2013, 15, 6102–6105 CrossRef CAS PubMed.
- G. Bringmann and D. Menche, Acc. Chem. Res., 2001, 34, 615–624 CrossRef CAS PubMed.
- (a) K.-S. Masters and S. Bräse, Angew. Chem., Int. Ed., 2013, 52, 866–869 CrossRef CAS PubMed; (b) K.-S. Masters, A. Bihlmeier, W. Klopper and S. Bräse, Chem.–Eur. J., 2013, 19, 17827–17835 CrossRef CAS PubMed.
- Charette and co-workers used pyridine as solvent (please see ref. 10a), which had been found to be optimal for our initial dioxepine work.
- (a) S. De, S. Misrah, B. N. Kakde, D. Dey and A. Bisai, J. Org. Chem., 2013, 78, 7823–7844 CrossRef CAS PubMed; (b) Reported nucleophilic aromatic substitution SNAr of aryl halide by NaOtBu under similar conditions: W. C. Wertjes, L. C. Wolfe, P. J. Waller and D. Kalyani, Org. Lett., 2013, 15, 5986–5989 CrossRef CAS PubMed; (c) The existence of both radical and nucleophilic substitution pathways at the one time under similar conditions is known. For a recent example, see H. Baars, A. Beyer, S. V. Kohlhepp and C. Bolm, Org. Lett., 2014, 16, 536–539 CrossRef CAS PubMed.
- (a) A. Baroudi, P. Flack and I. V. Alabugin, Chem.–Eur. J., 2010, 16, 12316–12320 CrossRef CAS PubMed; (b) A. Baroudi, J. Alicea and I. V. Alabugin, Chem.–Eur. J., 2010, 16, 7683–7687 CrossRef CAS PubMed; (c) A. Baroudi, J. Alicea, P. Flack, J. Kirincich and I. V. Alabugin, J. Org. Chem., 2011, 76, 1521–1537 CrossRef CAS PubMed.
- Comparison of arene C–H coupling partners used in this study with their boronic acid derivatives shows their relative costs; e.g. 1,4-dimethoxybenzene: AU$105/1000 g; 2,5-dimethoxyphenylboronic acid: AU$93/5 g (prices quoted online by Sigma-Aldrich).
- (a) W. C. Chen, Y. C. Hsu, W. C. Shih, C. Y. Lee, W. H. Chuang, Y. F. Tsai, P. P. Chen and T. G. Ong, Chem. Commun., 2012, 48, 6702–6704 RSC; (b) H. Zhang, R. Shi, A. Ding, L. Lu, B. Chen and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 12542–12545 CrossRef CAS PubMed; (c) H. Yi, A. Jutand and A. Lei, Chem. Commun., 2015, 51, 545–548 RSC.
- (a) A. B. Chopa, A. P. Murray and M. T. Lockhart, J. Organomet. Chem., 1999, 585, 35–42 CrossRef CAS; (b) N. Bodineau, J.-M. Mattalia, H. Hazimeh, K. L. Handoo, V. Timokhin, J.-C. Négrel and M. Chanon, Eur. J. Org. Chem., 2010, 13, 2476–2486 CrossRef; (c) G. S. Foray, A. B. Peňéňory' and R. A. Rossi, J. Phys. Org. Chem., 1995, 8, 356–358 CrossRef CAS.
- The results of Kwong and Lei (see ref. 5c) were supportive of a biarylation pathway for 1,4-diiodobenzene which involved a radical iodoarene anion intermediate.
- A. L. J. Beckwith and V. W. Bowry, J. Org. Chem., 1988, 53, 1632–1641 CrossRef CAS.
- Potassium dimsyl treatment of diamines makes superbases: E. M. Arnett and K. G. Venkatasubramaniam, J. Org. Chem., 1983, 48, 1569–1578 CrossRef CAS.
- (a) J. I. Brauman, N. J. Nelson and D. C. Kahl, J. Am. Chem. Soc., 1968, 90, 490–491 CrossRef CAS; (b) J. I. Brauman, N. J. Nelson and D. C. Kahl, J. Am. Chem. Soc., 1968, 90, 491–492 CrossRef CAS; (c) I. A. Romanskii, I. O. Shapiro and A. I. Shatenshtein, Reakts. Sposobn. Org. Soedin., 1968, 5, 452–455 CAS.
- J. Cuthbertson, V. J. Gray and J. D. Wilden, Chem. Commun., 2014, 50, 2575–2578 RSC.
- (a) J. H. Exner and E. C. Steiner, J. Am. Chem. Soc., 1974, 96, 1782–1787 CrossRef CAS; (b) E. M. Amett and K. G. Venkatasubramaniam, Tetrahedron Lett., 1981, 22, 987–990 CrossRef; (c) J. I. Brauman, J. A. Bryson, D. C. Kahl and N. J. Nelson, J. Am. Chem. Soc., 1970, 92, 6679–6680 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full 1H and 13C NMR spectra for 26 new compounds and a representation showing the set-up for coupling of solid at room-temperature arenes (Fig. S1) is compiled in a word document. See DOI: 10.1039/c5ra03460d |
| This journal is © The Royal Society of Chemistry 2015 |