
A strong charge-transfer effect in surface-enhanced Raman scattering induced by valence electrons of actinide elements†
 *ab
*ab
Abstract
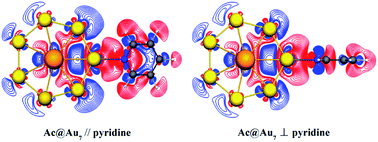
Surface-enhanced Raman scattering (SERS) is a powerful spectroscopic technique for highly sensitive molecular detection. As the great radial extent of 5f/6d orbitals is enough to support surface plasmons, SERS substrate materials containing actinide elements might bring a novel enhancement mechanism and even significantly enhanced magnitude. Here we study the SERS of a pyridine molecule on the typical actinide embedded structure Ac@Au7 and on a similar structure of pure gold Au8, respectively, using the time-dependent density functional theory (TDDFT) method. The calculated results show that the absorption spectrum of pyridine-Ac@Au7 at 456 nm presents a strong peak belonging to charge-transfer (CT) transition from the metal to the pyridine molecule. Surprisingly, the corresponding CT-SERS enhancement can reach an order of magnitude of 105, which could hardly be achieved in the pure gold systems (about 103–104). Furthermore, electronic structure analysis reveals that the initial orbitals of the CT transition contain a non-ignorable contribution from the 6d electrons of the Ac atom. Meanwhile, the contribution of the 6d electrons can be tuned by changing the conformation of the pyridine-Ac@Au7 complex. The current findings provide a theoretical basis for exploring and synthesizing SERS materials based on actinide elements, which might subsequently facilitate applications of such structures in nanostructural characterization, single-molecule SERS signal detections and biological molecular recognitions etc.
- This article is part of the themed collections: RSC Advances Editors' collection: f Block Chemistry and Surface enhanced Raman Spectroscopy: Editors collection for RSC Advances
 Please wait while we load your content...
Please wait while we load your content...

