
High selective fluorescence imaging of cesium distribution in Arabidopsis using a bis(trihydroxyphenyl)-appended fluorescent probe with a turn-on system†
Sung Ho Junga,
Tae Kyung Hyun*bc,
Jae-Yean Kimb and
Jong Hwa Jung*a
aDepartment of Chemistry and Research Institute of Natural Sciences, Gyeongsang National University, Jinju, South Korea. E-mail: jonghwa@gnu.ac.kr; Fax: +82-55-758-6027; Tel: +82-55-772-1488
bDivision of Applied Life Science (BK21plus), Plant Molecular Biology and Biotechnology Research Center, Gyeongsang National University, Jinju, South Korea
cDepartment of Industrial Plant Science & Technology, College of Agricultural, Life and Environmental Sciences, Chungbuk National University, Cheongju, South Korea
First published on 6th March 2015
Abstract
The bis(trihydroxyphenyl)-appended ligand 1 was found to display dramatically enhanced fluorescence upon the addition of Cs+, but not with any other metal ions. The self-assembled nanofiber of 1 was changed into a spherical structure upon addition of Cs+ in aqueous solution. Furthermore, 1 as a chemoprobe displayed high-resolution fluorescence imaging for cesium in Arabidopsis.
Cesium, a common contaminant in industrial, medical and nuclear wastes, can cause a number of negative health effects, including cardiovascular disease and gastrointestinal distress.1 Currently, there are several well-known detection methods for cesium in complex environmental and/or biological systems including atomic absorption spectroscopy (AAS), inductively coupled plasma mass spectroscopy (ICP-MS) and solid state sensors.2 Although these methods are highly sensitive and selective for cesium detection, they are often expensive, involve lengthy analysis time, and require sample destruction.
Very recently, the accident at the Fukushima Daiichi nuclear power plant in Japan released a large quantity of radioactive materials containing 137Cs into the atmosphere. In particular, 137Cs with a half-life of 30.17 years has the potential to be a long-term radioactive hazard detrimental to human health and agricultural production for generations.3 Furthermore, high concentrations of 137Cs remain in the vicinity of the meltdown site of the nuclear reactor, making a large area of land uninhabitable. Thus, detection methods for cesium that are easy and rapid are of great important in environmental and biological systems. For example, very recently, Dr K. Ariga et al. investigated on fluorescence imaging of cesium in Arabidopsis by using cesium green dye.4 The cesium green dye enabled high-resolution cesium imaging of cesium-containing particles and cesium contained in plants.4
Among a variety of detection methods, in particular, fluorimetric detection has recently attracted considerable attention for its high sensitivity and spatial resolution.5 However, only a few fluorescence-based methods have been reported for cesium detection, even though such methods have the potential to be both less expensive and non-destructive, and have been used successfully for the many analytes.6,15 For example, squaraine-based receptors have detected cesium with high affinities through a “turn-off” system.7 However, most of receptors have not yet been tested for their real application in living cell or plant tissues. Thus, the development of a selective fluorescent probe for Cs+ in plant tissues is desirable. Herein, we report a bis(trihydroxyphenyl)-appended chemoprobe for cesium in aqueous solution and in biological fluids.

The bis(trihydroxyphenyl)-based ligand 1 was synthesized by treating dipicolinic acid hydrazide with 2,3,4-trihydroxybenzaldehyde in ethanol using a slightly modified version of a previously reported8 procedure as shown in Scheme S1.† The desired product was a yellow color, and its structure was confirmed by 1H, 13C NMR, mass spectroscopy and elemental analysis.
We observed the effect of the addition of metal ions on the UV-Vis absorption and fluorescence spectra of 1 in aqueous solutions containing 1% DMF (v/v). The UV-Vis absorption spectrum of 1 (2.5 × 10−5 M) showed a maximum at 330 nm. The UV-Vis spectrum of 1 was gradually shifted to a longer wavelength upon addition of Cs2CO3, with the appearance of a new absorption maximum at 365 nm. In contrast, no significant absorption spectra changes were observed upon addition of other metal ions such as Li+, Na+, K+, Mg2+, Ca2+, Sr2+, Ag+, Zn2+ and Ni2+ (Fig. 1). These results indicate that Cs+ was selectively bound by 1 in aqueous solution. In addition, the oxygen atoms of both carbonyl groups and the oxygen ion atoms of the hydroxyl groups of 1 with deprotonation would act as binding sites for Cs+, which are well-known as a hard base.9
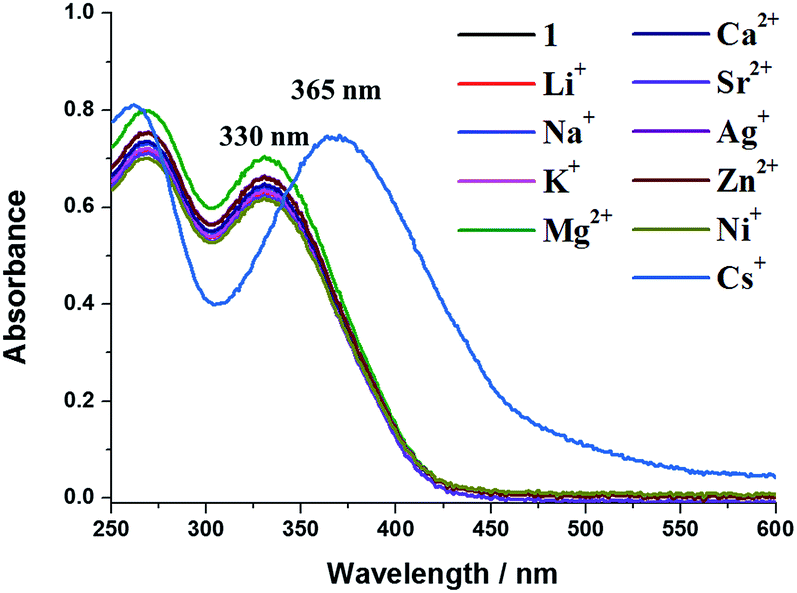 | ||
| Fig. 1 UV-Vis spectra of aggregated 1 (2.5 × 10−5 M) in the presence of various metal ions (5.0 × 10−5 M) in water–DMF mixture (99/1 v/v%). | ||
When ligand 1 (0.1 mM) was dissolved in a mixture of water–DMF (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v%), the solution of 1 was not transparent in the absence and the presence of Cs+ (10 equiv.). Thus, the SEM images of 1 without and with Cs+ in a mixture of water–DMF (99:1 v/v%) were observed by scanning electron microscopy (SEM). The SEM image of 1 without Cs+ in a mixture of water–DMF (99:1 v/v%) showed a fiber structure of 50 nm width and several micrometer lengths (Fig. 2A), which is attributed to the self-aggregation of 1 with the intermolecular hydrogen-bonding interaction and π–π stacking. In contrast, the SEM image of 1 with Cs+ (10 equiv.) showed nanoparticles of ca. 20 nm diameters (Fig. 2B). The morphological change of 1 is attributed to a geometric change of the ligand by complex formation with Cs+. The formation of nanoparticles of 1 with Cs+ was further confirmed by dynamic light scattering (DLS). As shown in Fig. S1,† the diameter distribution of the nanoparticles fell within the range of ca. 7–70 nm. On the other hand, no significant changes of morphologies of 1 were observed upon addition of Na2CO3 and K2CO3 (Fig. S2†), indicating that the aggregated 1 did not forms strong complex with Na2CO3 and K2CO3.
:1 v/v%), the solution of 1 was not transparent in the absence and the presence of Cs+ (10 equiv.). Thus, the SEM images of 1 without and with Cs+ in a mixture of water–DMF (99:1 v/v%) were observed by scanning electron microscopy (SEM). The SEM image of 1 without Cs+ in a mixture of water–DMF (99:1 v/v%) showed a fiber structure of 50 nm width and several micrometer lengths (Fig. 2A), which is attributed to the self-aggregation of 1 with the intermolecular hydrogen-bonding interaction and π–π stacking. In contrast, the SEM image of 1 with Cs+ (10 equiv.) showed nanoparticles of ca. 20 nm diameters (Fig. 2B). The morphological change of 1 is attributed to a geometric change of the ligand by complex formation with Cs+. The formation of nanoparticles of 1 with Cs+ was further confirmed by dynamic light scattering (DLS). As shown in Fig. S1,† the diameter distribution of the nanoparticles fell within the range of ca. 7–70 nm. On the other hand, no significant changes of morphologies of 1 were observed upon addition of Na2CO3 and K2CO3 (Fig. S2†), indicating that the aggregated 1 did not forms strong complex with Na2CO3 and K2CO3.
 | ||
| Fig. 2 SEM images of (A) aggregated 1 (0.1 mM) and (B) aggregated 1 (0.1 mM) + Cs+ (10.0 equiv.) in water–DMF mixture (99:1 v/v%). | ||
To investigate the selectivity for Cs+, we also observed the fluorescence changes of 1 upon addition of other metal ions in aqueous solution in the presence of 1 v/v% of DMF. Compound 1 exhibited extremely weak emission in the absence of Cs+ (excitation wavelength = 360 nm). As shown in Fig. 3A, 1 exhibited a strong emission upon addition of Cs2CO3 when the mixture was viewed under a UV lamp. The fluorescence intensity changes of 1, observed at 452 nm, increased quantitatively with the addition of increasing amounts of Cs+ (0–10 equiv.). Fluorescence enhancement of up to ∼200-fold was recorded with the addition of 10 equivalents of Cs+ (Fig. 3B). The significant fluorescence enhancement of 1 upon addition of Cs+ is due to aggregation-induced emission (AIE) effect (Fig. S3†).10 The ligand 1 in the absence of Cs+ aggregates with mainly π–π stacking between trihydroxyphenyl and trihydroxyphenyl groups with a linear structure to induce formation of a fibrillar structure, which is attributed to an aggregation-caused-quenching (ACQ) effect.11 The π–π stacking between the phenylene groups of 1 induced a quenching effect by H-aggregation mode.12 On the other hand, the addition of Cs+ induced formation of the pseudocyclic structure, which is an AIE effect. The addition of other metal ions, in contrast, had no effect on the fluorescence changes of 1. Thus, 1 responded selectively to Cs+, a response which can be attributed to the binding of Cs+ to 1. The addition of Cs+ resulted in a linear increase in the fluorescence intensities of the 1 (Fig. S4†), indicating that Cs+ became bound to 1. Compound 1 also showed excellent sensitivity with detection of 20 ppb of Cs+ (Fig. S5†), which is sensitive enough for its practical applications in measurements of Cs+ in biological fluids.
 | ||
| Fig. 3 (A) Photograph of aggregated 1 and aggregated 1 with Cs2(CO3) in water–DMF mixture (99:1 v/v%). (B) Fluorescence spectra of aggregated 1 (0.1 mM) in the presence of various metal ions (MNO3 or M2(NO3)2; 1.0 mM) in water–DMF mixture (99:1 v/v%). | ||
The UV-Vis spectra of 1 were also observed in a mixture of water and DMF (99:1 v/v) and pure DMF (Fig. S6A†). The UV-Vis spectrum of 1 in a mixture of water and DMF (99:1 v/v) exhibited absorbance in shorter wavelength than that of obtained in a pure DMF, suggesting that ligand 1 might be aggregated in a mixture of water and DMF (99:1 v/v). The fluorescence intensity of ligand 1 in pure DMF was smaller than that of obtained in a mixture of water and DMF (99:1 v/v) (Fig. S6B†), indicating that ligand 1 exists as monomeric species, not aggregation.
We examined the selectivity of aggregated 1 for Cs2CO3 with respect to alkali-metal carbonates such as K2CO3 and Na2CO3. The increases in fluorescence of 1 upon addition of Cs+ were higher than those observed with other alkali metal ions (Fig. S7†), suggesting that the aggregated 1–Cs+ complex was much more stable in comparison to the other alkali-metal carbonates.
We further investigated the interaction between 1 and Cs+ by using ESI-mass spectroscopy (Fig. S8†). The ESI-mass spectrum of 1 upon addition of Cs+ corresponded to the [1 + Cs]+ at m/z ∼ 600.67 while the peak at m/z ∼ 468.25 was related to 1, indicating that the binding between 1 and Cs+ led to a 1:1 stoichiometric ratio. The association constant (Ka) of 1 with Cs+ was calculated for 1:1 stoichiometry on the basis of a Benesie–Hildebrand plot,13 and it was calculated to be 1.18 × 103 M−1 (Fig. S9†).
To investigate the response time of 1 for sensing Cs+, the fluorescence spectra were recorded upon addition of Cs+ (0.1, 0.5 and 1.0 mM) to solutions of 1 (0.1 mM) and each solution was analyzed as a function of time. The interpretation of results revealed that after 60 min, the fluorescence intensity of any solution is independent of time; in other words, the sensing response time of 1 upon addition of Cs+ was constantly maintained within 60 min (Fig. S10†).
We considered that micrometer-scale imaging for Cs+ distribution could be extended to cellular-level imaging. Thus, we studied the practical applicability of 1 as an imaging probe for Cs+ to operate within living systems like plants. Nine-day-old Arabidopsis seedlings grown Murashige–Skoog (MS) medium containing 0.01 mM or 0.1 mM Cs2CO3 were vacuum infiltrated with 0.1 mM of the fluorescent probe 1, which revealed Cs+ content in the resulting plants of 22.8 nmol of Cs+ per mg of dry weight by elemental analysis (Fig. S8†), and the fluorescence signal was observed using a multiphoton laser scanning microscope. Upon a comparison between the fluorescence images of Cs+/probe 1-treated and Cs-treated plants, the high level of fluorescence signal was observed in vacuoles of leaf epidermal cells of Cs+/probe 1-treated plants but not of Cs-treated plants (Fig. 4). In addition, only probe-treated plants showed no detectable signal, indicating that endogenous autofluorescence arising from a variety of biomolecules such as flavins, fatty acids, cytochromes, chlorophylls, lignins, carotenoids and phenolic compounds have no or very little effect on probe 1-mediated fluorescence emission in plants.14 In higher plants, Cs+ influx to root cells occurs through K+/H+ symporters and voltage-insensitive cation channels, and is translocated to other organs like leaves and shoots.15 Although there is no known role for Cs in plant nutrition, excessive Cs is potentially toxic to plants. Therefore, accumulation of Cs in vacuole, which is consistent with previous observation using Cs fluorescent probe Cesium Green,4 indicates that vacuolar sequestration is potentially important mechanism for detoxification of Cs, suggesting potential application of probe 1 for studying the cellular mechanism for Cs detoxification in higher plants.
 | ||
| Fig. 4 Fluorescence images of plant grown of cotyledon of Arabidopsis (A) treated with aggregated 1 (0.1 mM) and absence of Cs2CO3; (B) treated with Cs2CO3 (0.1 mM) without the aggregated 1; (C) treated with Cs2CO3 (0.1 mM) and with aggregated 1 (0.1 mM). | ||
In conclusion, the bis(trihydroxyphenyl)-appended derivative 1 has been synthesized and its fluorogenic sensing properties have been studied. Ligand 1 alone was almost non-emissive, which is attributed to the aggregation-caused-quenching (ACQ) effect. In contrast, 1 exhibited a strong emission upon addition of cesium ion, due to the aggregation-induced emission (AIE) effect. Thus, 1 can function as a “turn-on” fluorescent chemoprobe for cesium ion. In particular, chemoprobe 1 displayed high-resolution fluorescence imaging for cesium in Arabidopsis. This result highlights the capability of chemoprobe 1 for visualization of cesium accumulated in plants at the organelle level. Thus, this method is potentially useful for detection of cesium contaminated by nuclear radiation and waste water in biological system.
Acknowledgements
This work was supported by a grant from the NRF (2014M2B2A9030338) supported from the Ministry of Education, Science and Technology, Korea. In addition, this work was partially supported by a grant from the Next-Generation BioGreen 21 Program (SSAC, grant#: PJ011177022015 and PJ009495), Rural Development Administration, Korea.Notes and references
- (a) A. M. Latifi, S. M. Nabavi, M. Mirzaei, M. Tavalaei, H. Ghafurian, C. Hellio and S. F. Nabavi, Toxicol. Environ. Chem., 2012, 94, 1670–1677 CrossRef CAS; (b) G. R. Peterson, Technol. Innovation, 2011, 13, 99–105 CrossRef CAS; (c) P. Melnikov and L. Z. Zanoni, Biol. Trace Elem. Res., 2010, 135, 1–99 CrossRef CAS PubMed.
- (a) K. Greda, P. Jamroz and P. Pohl, Talanta, 2013, 108, 74–82 CrossRef CAS PubMed; (b) M. Liezers, O. T. Farmer and M. L. Thomas, J. Radioanal. Nucl. Chem., 2009, 282, 309–313 CrossRef CAS; (c) T. Mori, M. Akamatsu, K. Okamoto, M. Sumita, Y. Tateyama, H. Sakai, J. P. Hill, M. Abe and K. Ariga, Sci. Technol. Adv. Mater., 2013, 14, 1–14 CrossRef.
- N. Kaneyasu, H. Ohashi, F. Suzuki, T. Okuda and F. Ikemori, Environ. Sci. Technol., 2012, 46, 5720–5726 CrossRef CAS PubMed.
- M. Akamatsu, H. Komatsu, T. Mori, E. Adams, R. Shin, H. Sakai, M. Abe, J. P. Hill and K. Ariga, ACS Appl. Mater. Interfaces, 2014, 6, 8208–8211 CAS.
- (a) X.-D. Wang and O. S. Wolfbeis, Chem. Soc. Rev., 2014, 43, 3666–3761 RSC; (b) S. Zhu, L. Ma, S. Wang, C. Chen, W. Zhang, L. Yang, W. Hang, J. P. Nolan, L. Wu and X. Yan, ACS Nano, 2014, 8, 10998–11006 CrossRef CAS PubMed; (c) Z. Yang, J. Cao, Y. He, J. H. Yang, T. Kim, X. Peng and J. S. Kim, Chem. Soc. Rev., 2014, 43, 4563–4601 RSC.
- (a) V. Souchon, I. Leray and B. Valeur, Chem. Commun., 2006, 4224–4226 RSC; (b) M. Shamsipur and H. R. Rahabi, Microchim. Acta, 2013, 180, 243–252 CrossRef CAS; (c) H. S. Seo, M. Mainul and S. H. Lee, J. Fluoresc., 2008, 18, 853–857 CrossRef CAS PubMed; (d) T. Mori, M. Akamatsu, K. Okamoto, M. Sumita, Y. Tateyama, H. Sakai, J. P. Hill, M. Abe and K. Ariga, Sci. Technol. Adv. Mater., 2013, 14, 015002 CrossRef; (e) H.-F. Ji, R. L. Hettich, G. M. Brown and R. Dabestani, J. Photochem. Photobiol., A, 1999, 70, 882–886 CrossRef CAS PubMed.
- B. Radaram, T. Mako and M. Levine, Dalton Trans., 2013, 42, 16276–16278 RSC.
- S. Chopra, N. Singh, P. Thangarasu, V. K. Bhardwaj and N. Kaur, Dyes Pigm., 2014, 106, 45–50 (J. Phys. Chem. B, 2008, 112, 14525–14529) CrossRef CAS PubMed.
- (a) J. L. Reed, Inorg. Chem., 2009, 48, 7151–7158 CrossRef CAS PubMed; (b) A. E. Rosamilia, F. Aricò and P. Tundo, J. Phys. Chem. B, 2008, 112, 14525–14529 CrossRef CAS PubMed; (c) R. G. Pearson and J. Songstad, J. Am. Chem. Soc., 1967, 89, 1827–1836 CrossRef CAS.
- (a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC; (b) R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494–4562 RSC; (c) Y. Gong, Y. Zhang, W. Z. Yuan, J. Z. Sun and Y. Zhang, J. Phys. Chem. C, 2014, 118, 10998–11005 CrossRef CAS; (d) P. Galer, R. C. Korošec, M. Vidmar and B. Šket, J. Am. Chem. Soc., 2014, 136, 7383–7394 CrossRef CAS PubMed; (e) J. Wang, J. Mei, R. Hu, J. Z. Sun, A. Qin and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 9956–9966 CrossRef CAS PubMed.
- (a) Y.-X. Li, M.-P. Pang, Z.-W. Zhang, G.-B. Lia and G.-X. Sun, RSC Adv., 2013, 3, 14950–14953 RSC; (b) W. Z. Yuan, P. Lu, S. Chen, J. W. Y. Lam, Z. Wang, Y. Liu, H. S. Kwok, Y. Ma and B. Z. Tang, Adv. Mater., 2010, 22, 2159–2163 CrossRef CAS PubMed; (c) A. Singh, S. Kaur, N. Singh and N. Kaur, Org. Biomol. Chem., 2014, 12, 2302–2309 RSC; (d) B. Wang and C. Yu, Angew. Chem., Int. Ed., 2010, 49, 1485–1488 CrossRef CAS PubMed; (e) H. Jiao, B. Wang, J. Chen, D. Liao, W. Li and C. Yu, Chem. Commun., 2012, 48, 7862–7864 RSC.
- S. H. Jung, K.-Y. Kwon and J. H. Jung, Chem. Commun., 2015, 51, 952–955 RSC.
- (a) H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS; (b) K. A. Conners, Binding Constants: The Measurement of Molecular Complex Stability, Wiley, New York, 1987 Search PubMed.
- L. Zhang, K. Gase, I. T. Baldwin and I. Gális, BioTechniques, 2010, 48, 125–133 CrossRef CAS PubMed.
- C. R. Hampton, M. R. Broadley and P. J. White, Nukleonika, 2005, 50, S3–S8 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectroscopic analysis. See DOI: 10.1039/c5ra03371c |
| This journal is © The Royal Society of Chemistry 2015 |